 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeNo Foundations without Foundations -- Why semi-mechanistic models are essential for regulatory biology
Jan 31, 2025Despite substantial efforts, deep learning has not yet delivered a transformative impact on elucidating regulatory biology, particularly in the realm of predicting gene expression profiles. Here, we argue that genuine "foundation models" of regulatory biology will remain out of reach unless guided by frameworks that integrate mechanistic insight with principled experimental design. We present one such ground-up, semi-mechanistic framework that unifies perturbation-based experimental designs across both in vitro and in vivo CRISPR screens, accounting for differentiating and non-differentiating cellular systems. By revealing previously unrecognised assumptions in published machine learning methods, our approach clarifies links with popular techniques such as variational autoencoders and structural causal models. In practice, this framework suggests a modified loss function that we demonstrate can improve predictive performance, and further suggests an error analysis that informs batching strategies. Ultimately, since cellular regulation emerges from innumerable interactions amongst largely uncharted molecular components, we contend that systems-level understanding cannot be achieved through structural biology alone. Instead, we argue that real progress will require a first-principles perspective on how experiments capture biological phenomena, how data are generated, and how these processes can be reflected in more faithful modelling architectures.
BioNeMo Framework: a modular, high-performance library for AI model development in drug discovery
Nov 15, 2024



Artificial Intelligence models encoding biology and chemistry are opening new routes to high-throughput and high-quality in-silico drug development. However, their training increasingly relies on computational scale, with recent protein language models (pLM) training on hundreds of graphical processing units (GPUs). We introduce the BioNeMo Framework to facilitate the training of computational biology and chemistry AI models across hundreds of GPUs. Its modular design allows the integration of individual components, such as data loaders, into existing workflows and is open to community contributions. We detail technical features of the BioNeMo Framework through use cases such as pLM pre-training and fine-tuning. On 256 NVIDIA A100s, BioNeMo Framework trains a three billion parameter BERT-based pLM on over one trillion tokens in 4.2 days. The BioNeMo Framework is open-source and free for everyone to use.
Season combinatorial intervention predictions with Salt & Peper
Apr 25, 2024



Interventions play a pivotal role in the study of complex biological systems. In drug discovery, genetic interventions (such as CRISPR base editing) have become central to both identifying potential therapeutic targets and understanding a drug's mechanism of action. With the advancement of CRISPR and the proliferation of genome-scale analyses such as transcriptomics, a new challenge is to navigate the vast combinatorial space of concurrent genetic interventions. Addressing this, our work concentrates on estimating the effects of pairwise genetic combinations on the cellular transcriptome. We introduce two novel contributions: Salt, a biologically-inspired baseline that posits the mostly additive nature of combination effects, and Peper, a deep learning model that extends Salt's additive assumption to achieve unprecedented accuracy. Our comprehensive comparison against existing state-of-the-art methods, grounded in diverse metrics, and our out-of-distribution analysis highlight the limitations of current models in realistic settings. This analysis underscores the necessity for improved modelling techniques and data acquisition strategies, paving the way for more effective exploration of genetic intervention effects.
PyRelationAL: A Library for Active Learning Research and Development
May 23, 2022
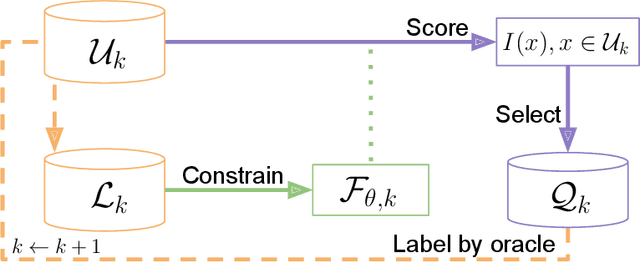
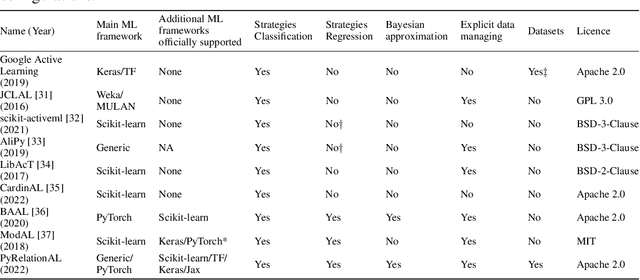
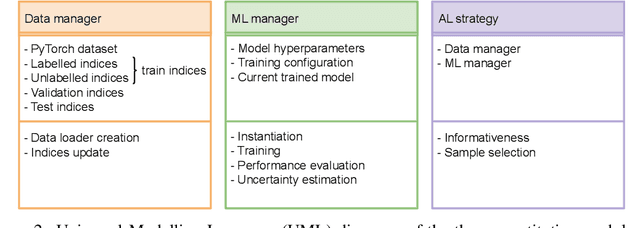
In constrained real-world scenarios where it is challenging or costly to generate data, disciplined methods for acquiring informative new data points are of fundamental importance for the efficient training of machine learning (ML) models. Active learning (AL) is a subfield of ML focused on the development of methods to iteratively and economically acquire data through strategically querying new data points that are the most useful for a particular task. Here, we introduce PyRelationAL, an open source library for AL research. We describe a modular toolkit that is compatible with diverse ML frameworks (e.g. PyTorch, Scikit-Learn, TensorFlow, JAX). Furthermore, to help accelerate research and development in the field, the library implements a number of published methods and provides API access to wide-ranging benchmark datasets and AL task configurations based on existing literature. The library is supplemented by an expansive set of tutorials, demos, and documentation to help users get started. We perform experiments on the PyRelationAL collection of benchmark datasets and showcase the considerable economies that AL can provide. PyRelationAL is maintained using modern software engineering practices - with an inclusive contributor code of conduct - to promote long term library quality and utilisation.
RECOVER: sequential model optimization platform for combination drug repurposing identifies novel synergistic compounds in vitro
Feb 07, 2022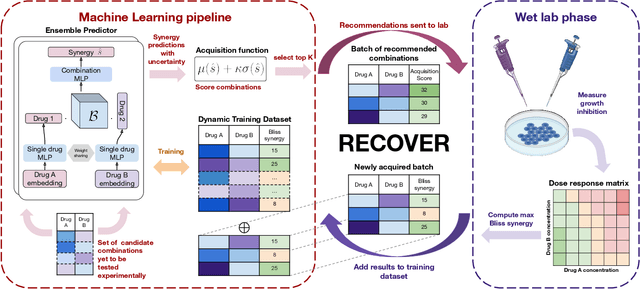

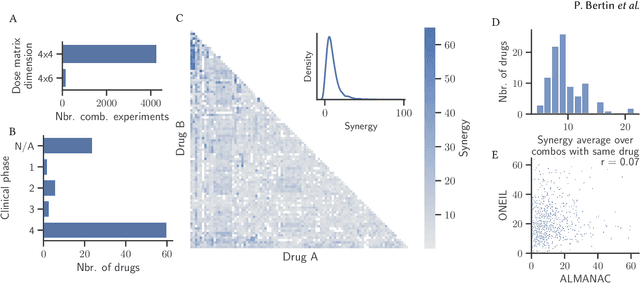
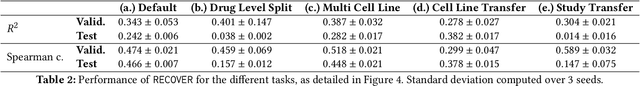
Selecting optimal drug repurposing combinations for further preclinical development is a challenging technical feat. Due to the toxicity of many therapeutic agents (e.g., chemotherapy), practitioners have favoured selection of synergistic compounds whereby lower doses can be used whilst maintaining high efficacy. For a fixed small molecule library, an exhaustive combinatorial chemical screen becomes infeasible to perform for academic and industry laboratories alike. Deep learning models have achieved state-of-the-art results in silico for the prediction of synergy scores. However, databases of drug combinations are highly biased towards synergistic agents and these results do not necessarily generalise out of distribution. We employ a sequential model optimization search applied to a deep learning model to quickly discover highly synergistic drug combinations active against a cancer cell line, while requiring substantially less screening than an exhaustive evaluation. Through iteratively adapting the model to newly acquired data, after only 3 rounds of ML-guided experimentation (including a calibration round), we find that the set of combinations queried by our model is enriched for highly synergistic combinations. Remarkably, we rediscovered a synergistic drug combination that was later confirmed to be under study within clinical trials.
Utilising Graph Machine Learning within Drug Discovery and Development
Dec 09, 2020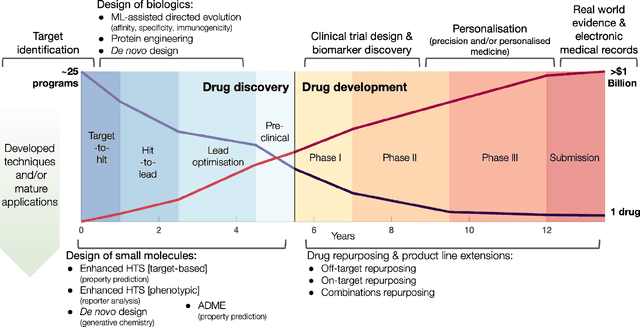
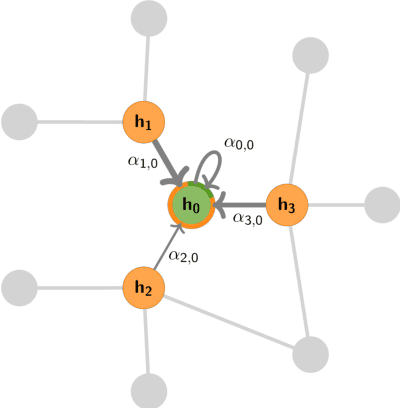
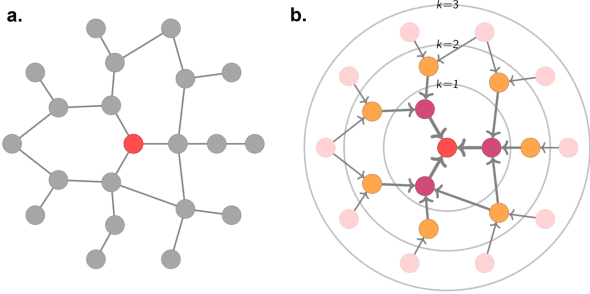
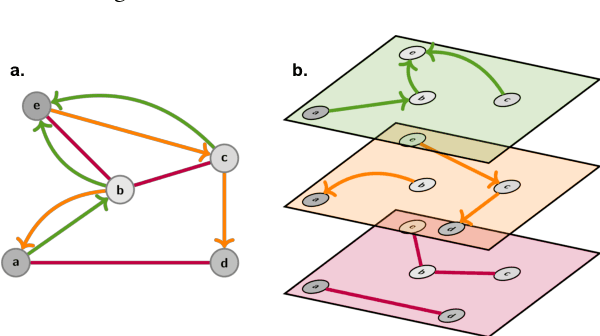
Graph Machine Learning (GML) is receiving growing interest within the pharmaceutical and biotechnology industries for its ability to model biomolecular structures, the functional relationships between them, and integrate multi-omic datasets - amongst other data types. Herein, we present a multidisciplinary academic-industrial review of the topic within the context of drug discovery and development. After introducing key terms and modelling approaches, we move chronologically through the drug development pipeline to identify and summarise work incorporating: target identification, design of small molecules and biologics, and drug repurposing. Whilst the field is still emerging, key milestones including repurposed drugs entering in vivo studies, suggest graph machine learning will become a modelling framework of choice within biomedical machine learning.