 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA Pathology Foundation Model for Gastric Cancer with Real-World Validation
Jun 03, 2026Gastric cancer remains a major cause of cancer mortality, yet its histological and molecular heterogeneity complicates diagnosis and risk stratification. General-purpose pathology foundation models (PFMs) often plateau on fine-grained endpoints central to gastric cancer care, and few have undergone rigorous prospective validation or clinical reader studies. We present GRACE, a Gastric-specific foundation model for Real-world Assessment and Clinical dEcision support. GRACE was developed from multicenter gastric pathology datasets totaling 48,364 primarily HE-stained whole-slide images from 37,493 patients. When evaluated on 28 clinically relevant tasks, GRACE consistently outperformed representative pancancer PFMs, achieving a macro-AUC of 0.9188, with strong performance for precancerous lesion diagnosis (macro-AUC 0.9322), tumor histopathological assessment (macro-AUC 0.9119), molecular profiling (macro-AUC 0.8682), and prognostic prediction. Beyond benchmarking, GRACE's translational value was substantiated through a rigorous evidence chain. Under safety-gated criteria requiring 100% NPV for rule-out and 100% PPV for rule-in, GRACE streamlined review for up to 69.6% of malignancy-diagnosis cases and triaged 46.8% of MMR-IHC follow-up requests. This translational feasibility was further strengthened by a randomized crossover reader study of pathologist-AI collaboration. With GRACE assistance, diagnostic accuracy improved from 82.0% to 89.9%, yielding nearly twofold higher adjusted odds of a correct diagnosis (OR 1.987) alongside concurrent gains in sensitivity and specificity. AI assistance also reduced diagnostic time by 14.9%, elevated diagnostic confidence by 9.0%, and markedly improved inter-rater agreement. When calibrated to maintain non-inferior performance to senior pathologists, the AI-assisted workflow could triage 60.7% of atrophy and 82.7% of intestinal metaplasia cases.
Spatial Transcriptomics-Guided Alignment Enhances Molecular Profiling in Pathology Foundation Model
May 29, 2026Comprehensive molecular profiling is essential for modern precision oncology but remains hindered by prohibitive costs, specimen exhaustion, and protracted turnaround times. While pathology foundation models (PFMs) have demonstrated potential for inferring molecular phenotypes from routine hematoxylin and eosin (H&E) whole-slide images (WSIs), current architectures primarily rely on vision-centric self-supervised learning or vision-language alignment, lacking the spatially resolved molecular supervision required to connect subtle morphological features with underlying genomic alterations. Spatial transcriptomics (ST) emerges as a transformative technology that enables transcriptomic quantification within intact tissue sections, thereby preserving the precise spatial link between histology and molecular profiles. In this study, we present a Spatial Transcriptomics-guided Alignment framework for Molecular Profiling (STAMP), which endows PFMs with intrinsic molecular awareness. To support this paradigm, we curated HumanST-1k, a human ST dataset spanning diverse anatomical organs and sequencing platforms. This atlas yields 1.8 million pairs of H&E patches and corresponding transcriptomic profiles, providing a corpus that links histological structures with their molecular states. To mitigate the technical noise inherent to raw transcriptomics, STAMP applies a pathway-informed alignment strategy that aggregates transcriptomic data into biologically functional pathways, which are subsequently integrated into PFMs via parameter-efficient fine-tuning. This alignment enriches the representation space of PFMs and unlocks their capacity to resolve sub-visual molecular signatures. The clinical utility of these augmented representations was validated through a multi-tier evaluation framework.
A Clinically Validated Foundation Model for Comprehensive Lung Pathology Interpretation
May 25, 2026Pathological assessment guides lung cancer diagnosis, treatment selection, and prognostic evaluation, yet current CPath approaches rely on task-specific models for isolated objectives. Although pan-cancer foundation models offer versatility, they lack subspecialty-level depth and have not been evaluated across clinical workflows or prospectively validated in real-world settings. We introduce PulmoFoundation, a multi-center, prospectively validated, randomized controlled trial (RCT)-evaluated foundation model for comprehensive lung pathology assessment across pre-operative, intra-operative, and post-operative care. Built upon Virchow2 via subspecialty-specific pretraining using ~40,000 diagnostic H&E-stained whole-slide images (WSIs), PulmoFoundation was systematically evaluated on ~26,000 WSIs across 32 clinically relevant tasks. In addition to accurately predicting molecular markers and patient survival, our model achieves clinical-grade performance in core diagnostic tasks across biopsy, frozen section, and surgical resection slides. In a registered prospective study of 1,357 patients across 11 diagnostic tasks, our model achieved an average AUC of 92.3%. Using pre-specified triage thresholds, PulmoFoundation could reduce additional second-review burden for 68.8% of biopsies and 83.0% of frozen sections, and defer 44.5% of IHC stain orders, with PPVs of 1.0, 0.991, and 0.966. Beyond prospective validation, we conducted a crossover RCT with eight pathologists, in which AI assistance improved diagnostic accuracy across 4,928 case-reader pairs (91.7% w/ AI vs. 83.8% w/o AI). AI assistance also reduced median diagnostic time by 19.6%, increased diagnostic confidence by 8.7%, and improved inter-rater agreement from moderate (kappa = 0.56) to substantial (kappa = 0.76). Together, these evaluations support PulmoFoundation as a clinically validated decision-support system for lung pathology.
A Deployment-Friendly Foundational Framework for Efficient Computational Pathology
Feb 15, 2026Pathology foundation models (PFMs) have enabled robust generalization in computational pathology through large-scale datasets and expansive architectures, but their substantial computational cost, particularly for gigapixel whole slide images, limits clinical accessibility and scalability. Here, we present LitePath, a deployment-friendly foundational framework designed to mitigate model over-parameterization and patch level redundancy. LitePath integrates LiteFM, a compact model distilled from three large PFMs (Virchow2, H-Optimus-1 and UNI2) using 190 million patches, and the Adaptive Patch Selector (APS), a lightweight component for task-specific patch selection. The framework reduces model parameters by 28x and lowers FLOPs by 403.5x relative to Virchow2, enabling deployment on low-power edge hardware such as the NVIDIA Jetson Orin Nano Super. On this device, LitePath processes 208 slides per hour, 104.5x faster than Virchow2, and consumes 0.36 kWh per 3,000 slides, 171x lower than Virchow2 on an RTX3090 GPU. We validated accuracy using 37 cohorts across four organs and 26 tasks (26 internal, 9 external, and 2 prospective), comprising 15,672 slides from 9,808 patients disjoint from the pretraining data. LitePath ranks second among 19 evaluated models and outperforms larger models including H-Optimus-1, mSTAR, UNI2 and GPFM, while retaining 99.71% of the AUC of Virchow2 on average. To quantify the balance between accuracy and efficiency, we propose the Deployability Score (D-Score), defined as the weighted geometric mean of normalized AUC and normalized FLOP, where LitePath achieves the highest value, surpassing Virchow2 by 10.64%. These results demonstrate that LitePath enables rapid, cost-effective and energy-efficient pathology image analysis on accessible hardware while maintaining accuracy comparable to state-of-the-art PFMs and reducing the carbon footprint of AI deployment.
MambaMIL+: Modeling Long-Term Contextual Patterns for Gigapixel Whole Slide Image
Dec 19, 2025



Whole-slide images (WSIs) are an important data modality in computational pathology, yet their gigapixel resolution and lack of fine-grained annotations challenge conventional deep learning models. Multiple instance learning (MIL) offers a solution by treating each WSI as a bag of patch-level instances, but effectively modeling ultra-long sequences with rich spatial context remains difficult. Recently, Mamba has emerged as a promising alternative for long sequence learning, scaling linearly to thousands of tokens. However, despite its efficiency, it still suffers from limited spatial context modeling and memory decay, constraining its effectiveness to WSI analysis. To address these limitations, we propose MambaMIL+, a new MIL framework that explicitly integrates spatial context while maintaining long-range dependency modeling without memory forgetting. Specifically, MambaMIL+ introduces 1) overlapping scanning, which restructures the patch sequence to embed spatial continuity and instance correlations; 2) a selective stripe position encoder (S2PE) that encodes positional information while mitigating the biases of fixed scanning orders; and 3) a contextual token selection (CTS) mechanism, which leverages supervisory knowledge to dynamically enlarge the contextual memory for stable long-range modeling. Extensive experiments on 20 benchmarks across diagnostic classification, molecular prediction, and survival analysis demonstrate that MambaMIL+ consistently achieves state-of-the-art performance under three feature extractors (ResNet-50, PLIP, and CONCH), highlighting its effectiveness and robustness for large-scale computational pathology
A Clinical-grade Universal Foundation Model for Intraoperative Pathology
Oct 06, 2025Intraoperative pathology is pivotal to precision surgery, yet its clinical impact is constrained by diagnostic complexity and the limited availability of high-quality frozen-section data. While computational pathology has made significant strides, the lack of large-scale, prospective validation has impeded its routine adoption in surgical workflows. Here, we introduce CRISP, a clinical-grade foundation model developed on over 100,000 frozen sections from eight medical centers, specifically designed to provide Clinical-grade Robust Intraoperative Support for Pathology (CRISP). CRISP was comprehensively evaluated on more than 15,000 intraoperative slides across nearly 100 retrospective diagnostic tasks, including benign-malignant discrimination, key intraoperative decision-making, and pan-cancer detection, etc. The model demonstrated robust generalization across diverse institutions, tumor types, and anatomical sites-including previously unseen sites and rare cancers. In a prospective cohort of over 2,000 patients, CRISP sustained high diagnostic accuracy under real-world conditions, directly informing surgical decisions in 92.6% of cases. Human-AI collaboration further reduced diagnostic workload by 35%, avoided 105 ancillary tests and enhanced detection of micrometastases with 87.5% accuracy. Together, these findings position CRISP as a clinical-grade paradigm for AI-driven intraoperative pathology, bridging computational advances with surgical precision and accelerating the translation of artificial intelligence into routine clinical practice.
A Multimodal Foundation Model to Enhance Generalizability and Data Efficiency for Pan-cancer Prognosis Prediction
Sep 16, 2025Multimodal data provides heterogeneous information for a holistic understanding of the tumor microenvironment. However, existing AI models often struggle to harness the rich information within multimodal data and extract poorly generalizable representations. Here we present MICE (Multimodal data Integration via Collaborative Experts), a multimodal foundation model that effectively integrates pathology images, clinical reports, and genomics data for precise pan-cancer prognosis prediction. Instead of conventional multi-expert modules, MICE employs multiple functionally diverse experts to comprehensively capture both cross-cancer and cancer-specific insights. Leveraging data from 11,799 patients across 30 cancer types, we enhanced MICE's generalizability by coupling contrastive and supervised learning. MICE outperformed both unimodal and state-of-the-art multi-expert-based multimodal models, demonstrating substantial improvements in C-index ranging from 3.8% to 11.2% on internal cohorts and 5.8% to 8.8% on independent cohorts, respectively. Moreover, it exhibited remarkable data efficiency across diverse clinical scenarios. With its enhanced generalizability and data efficiency, MICE establishes an effective and scalable foundation for pan-cancer prognosis prediction, holding strong potential to personalize tailored therapies and improve treatment outcomes.
A Versatile Pathology Co-pilot via Reasoning Enhanced Multimodal Large Language Model
Jul 23, 2025



Multimodal large language models (MLLMs) have emerged as powerful tools for computational pathology, offering unprecedented opportunities to integrate pathological images with language context for comprehensive diagnostic analysis. These models hold particular promise for automating complex tasks that traditionally require expert interpretation of pathologists. However, current MLLM approaches in pathology demonstrate significantly constrained reasoning capabilities, primarily due to their reliance on expensive chain-of-thought annotations. Additionally, existing methods remain limited to simplex application of visual question answering (VQA) at region-of-interest (ROI) level, failing to address the full spectrum of diagnostic needs such as ROI classification, detection, segmentation, whole-slide-image (WSI) classification and VQA in clinical practice. In this study, we present SmartPath-R1, a versatile MLLM capable of simultaneously addressing both ROI-level and WSI-level tasks while demonstrating robust pathological reasoning capability. Our framework combines scale-dependent supervised fine-tuning and task-aware reinforcement fine-tuning, which circumvents the requirement for chain-of-thought supervision by leveraging the intrinsic knowledge within MLLM. Furthermore, SmartPath-R1 integrates multiscale and multitask analysis through a mixture-of-experts mechanism, enabling dynamic processing for diverse tasks. We curate a large-scale dataset comprising 2.3M ROI samples and 188K WSI samples for training and evaluation. Extensive experiments across 72 tasks validate the effectiveness and superiority of the proposed approach. This work represents a significant step toward developing versatile, reasoning-enhanced AI systems for precision pathology.
Genome-Anchored Foundation Model Embeddings Improve Molecular Prediction from Histology Images
Jun 24, 2025

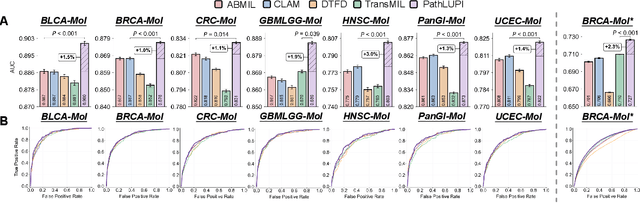

Precision oncology requires accurate molecular insights, yet obtaining these directly from genomics is costly and time-consuming for broad clinical use. Predicting complex molecular features and patient prognosis directly from routine whole-slide images (WSI) remains a major challenge for current deep learning methods. Here we introduce PathLUPI, which uses transcriptomic privileged information during training to extract genome-anchored histological embeddings, enabling effective molecular prediction using only WSIs at inference. Through extensive evaluation across 49 molecular oncology tasks using 11,257 cases among 20 cohorts, PathLUPI demonstrated superior performance compared to conventional methods trained solely on WSIs. Crucially, it achieves AUC $\geq$ 0.80 in 14 of the biomarker prediction and molecular subtyping tasks and C-index $\geq$ 0.70 in survival cohorts of 5 major cancer types. Moreover, PathLUPI embeddings reveal distinct cellular morphological signatures associated with specific genotypes and related biological pathways within WSIs. By effectively encoding molecular context to refine WSI representations, PathLUPI overcomes a key limitation of existing models and offers a novel strategy to bridge molecular insights with routine pathology workflows for wider clinical application.
PathBench: A comprehensive comparison benchmark for pathology foundation models towards precision oncology
May 26, 2025



The emergence of pathology foundation models has revolutionized computational histopathology, enabling highly accurate, generalized whole-slide image analysis for improved cancer diagnosis, and prognosis assessment. While these models show remarkable potential across cancer diagnostics and prognostics, their clinical translation faces critical challenges including variability in optimal model across cancer types, potential data leakage in evaluation, and lack of standardized benchmarks. Without rigorous, unbiased evaluation, even the most advanced PFMs risk remaining confined to research settings, delaying their life-saving applications. Existing benchmarking efforts remain limited by narrow cancer-type focus, potential pretraining data overlaps, or incomplete task coverage. We present PathBench, the first comprehensive benchmark addressing these gaps through: multi-center in-hourse datasets spanning common cancers with rigorous leakage prevention, evaluation across the full clinical spectrum from diagnosis to prognosis, and an automated leaderboard system for continuous model assessment. Our framework incorporates large-scale data, enabling objective comparison of PFMs while reflecting real-world clinical complexity. All evaluation data comes from private medical providers, with strict exclusion of any pretraining usage to avoid data leakage risks. We have collected 15,888 WSIs from 8,549 patients across 10 hospitals, encompassing over 64 diagnosis and prognosis tasks. Currently, our evaluation of 19 PFMs shows that Virchow2 and H-Optimus-1 are the most effective models overall. This work provides researchers with a robust platform for model development and offers clinicians actionable insights into PFM performance across diverse clinical scenarios, ultimately accelerating the translation of these transformative technologies into routine pathology practice.