 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMolecular Dynamics
Papers and Code
Root Cause Analysis of Hydrogen Bond Separation in Spatio-Temporal Molecular Dynamics using Causal Models
Aug 17, 2025



Molecular dynamics simulations (MDS) face challenges, including resource-heavy computations and the need to manually scan outputs to detect "interesting events," such as the formation and persistence of hydrogen bonds between atoms of different molecules. A critical research gap lies in identifying the underlying causes of hydrogen bond formation and separation -understanding which interactions or prior events contribute to their emergence over time. With this challenge in mind, we propose leveraging spatio-temporal data analytics and machine learning models to enhance the detection of these phenomena. In this paper, our approach is inspired by causal modeling and aims to identify the root cause variables of hydrogen bond formation and separation events. Specifically, we treat the separation of hydrogen bonds as an "intervention" occurring and represent the causal structure of the bonding and separation events in the MDS as graphical causal models. These causal models are built using a variational autoencoder-inspired architecture that enables us to infer causal relationships across samples with diverse underlying causal graphs while leveraging shared dynamic information. We further include a step to infer the root causes of changes in the joint distribution of the causal models. By constructing causal models that capture shifts in the conditional distributions of molecular interactions during bond formation or separation, this framework provides a novel perspective on root cause analysis in molecular dynamic systems. We validate the efficacy of our model empirically on the atomic trajectories that used MDS for chiral separation, demonstrating that we can predict many steps in the future and also find the variables driving the observed changes in the system.
Phenome-Wide Multi-Omics Integration Uncovers Distinct Archetypes of Human Aging
Oct 14, 2025Aging is a highly complex and heterogeneous process that progresses at different rates across individuals, making biological age (BA) a more accurate indicator of physiological decline than chronological age. While previous studies have built aging clocks using single-omics data, they often fail to capture the full molecular complexity of human aging. In this work, we leveraged the Human Phenotype Project, a large-scale cohort of 12,000 adults aged 30--70 years, with extensive longitudinal profiling that includes clinical, behavioral, environmental, and multi-omics datasets -- spanning transcriptomics, lipidomics, metabolomics, and the microbiome. By employing advanced machine learning frameworks capable of modeling nonlinear biological dynamics, we developed and rigorously validated a multi-omics aging clock that robustly predicts diverse health outcomes and future disease risk. Unsupervised clustering of the integrated molecular profiles from multi-omics uncovered distinct biological subtypes of aging, revealing striking heterogeneity in aging trajectories and pinpointing pathway-specific alterations associated with different aging patterns. These findings demonstrate the power of multi-omics integration to decode the molecular landscape of aging and lay the groundwork for personalized healthspan monitoring and precision strategies to prevent age-related diseases.
Amortized Sampling with Transferable Normalizing Flows
Aug 25, 2025Efficient equilibrium sampling of molecular conformations remains a core challenge in computational chemistry and statistical inference. Classical approaches such as molecular dynamics or Markov chain Monte Carlo inherently lack amortization; the computational cost of sampling must be paid in-full for each system of interest. The widespread success of generative models has inspired interest into overcoming this limitation through learning sampling algorithms. Despite performing on par with conventional methods when trained on a single system, learned samplers have so far demonstrated limited ability to transfer across systems. We prove that deep learning enables the design of scalable and transferable samplers by introducing Prose, a 280 million parameter all-atom transferable normalizing flow trained on a corpus of peptide molecular dynamics trajectories up to 8 residues in length. Prose draws zero-shot uncorrelated proposal samples for arbitrary peptide systems, achieving the previously intractable transferability across sequence length, whilst retaining the efficient likelihood evaluation of normalizing flows. Through extensive empirical evaluation we demonstrate the efficacy of Prose as a proposal for a variety of sampling algorithms, finding a simple importance sampling-based finetuning procedure to achieve superior performance to established methods such as sequential Monte Carlo on unseen tetrapeptides. We open-source the Prose codebase, model weights, and training dataset, to further stimulate research into amortized sampling methods and finetuning objectives.
LeMat-Traj: A Scalable and Unified Dataset of Materials Trajectories for Atomistic Modeling
Aug 28, 2025



The development of accurate machine learning interatomic potentials (MLIPs) is limited by the fragmented availability and inconsistent formatting of quantum mechanical trajectory datasets derived from Density Functional Theory (DFT). These datasets are expensive to generate yet difficult to combine due to variations in format, metadata, and accessibility. To address this, we introduce LeMat-Traj, a curated dataset comprising over 120 million atomic configurations aggregated from large-scale repositories, including the Materials Project, Alexandria, and OQMD. LeMat-Traj standardizes data representation, harmonizes results and filters for high-quality configurations across widely used DFT functionals (PBE, PBESol, SCAN, r2SCAN). It significantly lowers the barrier for training transferrable and accurate MLIPs. LeMat-Traj spans both relaxed low-energy states and high-energy, high-force structures, complementing molecular dynamics and active learning datasets. By fine-tuning models pre-trained on high-force data with LeMat-Traj, we achieve a significant reduction in force prediction errors on relaxation tasks. We also present LeMaterial-Fetcher, a modular and extensible open-source library developed for this work, designed to provide a reproducible framework for the community to easily incorporate new data sources and ensure the continued evolution of large-scale materials datasets. LeMat-Traj and LeMaterial-Fetcher are publicly available at https://huggingface.co/datasets/LeMaterial/LeMat-Traj and https://github.com/LeMaterial/lematerial-fetcher.
Probing the Critical Point (CritPt) of AI Reasoning: a Frontier Physics Research Benchmark
Oct 01, 2025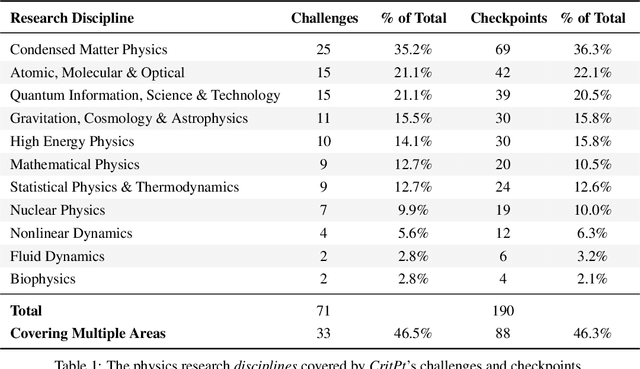



While large language models (LLMs) with reasoning capabilities are progressing rapidly on high-school math competitions and coding, can they reason effectively through complex, open-ended challenges found in frontier physics research? And crucially, what kinds of reasoning tasks do physicists want LLMs to assist with? To address these questions, we present the CritPt (Complex Research using Integrated Thinking - Physics Test, pronounced "critical point"), the first benchmark designed to test LLMs on unpublished, research-level reasoning tasks that broadly covers modern physics research areas, including condensed matter, quantum physics, atomic, molecular & optical physics, astrophysics, high energy physics, mathematical physics, statistical physics, nuclear physics, nonlinear dynamics, fluid dynamics and biophysics. CritPt consists of 71 composite research challenges designed to simulate full-scale research projects at the entry level, which are also decomposed to 190 simpler checkpoint tasks for more fine-grained insights. All problems are newly created by 50+ active physics researchers based on their own research. Every problem is hand-curated to admit a guess-resistant and machine-verifiable answer and is evaluated by an automated grading pipeline heavily customized for advanced physics-specific output formats. We find that while current state-of-the-art LLMs show early promise on isolated checkpoints, they remain far from being able to reliably solve full research-scale challenges: the best average accuracy among base models is only 4.0% , achieved by GPT-5 (high), moderately rising to around 10% when equipped with coding tools. Through the realistic yet standardized evaluation offered by CritPt, we highlight a large disconnect between current model capabilities and realistic physics research demands, offering a foundation to guide the development of scientifically grounded AI tools.
Artificial Intelligence Virtual Cells: From Measurements to Decisions across Modality, Scale, Dynamics, and Evaluation
Oct 14, 2025Artificial Intelligence Virtual Cells (AIVCs) aim to learn executable, decision-relevant models of cell state from multimodal, multiscale measurements. Recent studies have introduced single-cell and spatial foundation models, improved cross-modality alignment, scaled perturbation atlases, and explored pathway-level readouts. Nevertheless, although held-out validation is standard practice, evaluations remain predominantly within single datasets and settings; evidence indicates that transport across laboratories and platforms is often limited, that some data splits are vulnerable to leakage and coverage bias, and that dose, time and combination effects are not yet systematically handled. Cross-scale coupling also remains constrained, as anchors linking molecular, cellular and tissue levels are sparse, and alignment to scientific or clinical readouts varies across studies. We propose a model-agnostic Cell-State Latent (CSL) perspective that organizes learning via an operator grammar: measurement, lift/project for cross-scale coupling, and intervention for dosing and scheduling. This view motivates a decision-aligned evaluation blueprint across modality, scale, context and intervention, and emphasizes function-space readouts such as pathway activity, spatial neighborhoods and clinically relevant endpoints. We recommend operator-aware data design, leakage-resistant partitions, and transparent calibration and reporting to enable reproducible, like-for-like comparisons.
Machine Learning Workflow for Analysis of High-Dimensional Order Parameter Space: A Case Study of Polymer Crystallization from Molecular Dynamics Simulations
Jul 23, 2025



Currently, identification of crystallization pathways in polymers is being carried out using molecular simulation-based data on a preset cut-off point on a single order parameter (OP) to define nucleated or crystallized regions. Aside from sensitivity to cut-off, each of these OPs introduces its own systematic biases. In this study, an integrated machine learning workflow is presented to accurately quantify crystallinity in polymeric systems using atomistic molecular dynamics data. Each atom is represented by a high-dimensional feature vector that combines geometric, thermodynamic-like, and symmetry-based descriptors. Low dimensional embeddings are employed to expose latent structural fingerprints within atomic environments. Subsequently, unsupervised clustering on the embeddings identified crystalline and amorphous atoms with high fidelity. After generating high quality labels with multidimensional data, we use supervised learning techniques to identify a minimal set of order parameters that can fully capture this label. Various tests were conducted to reduce the feature set, demonstrating that using only three order parameters is sufficient to recreate the crystallization labels. Based on these observed OPs, the crystallinity index (C-index) is defined as the logistic regression model's probability of crystallinity, remaining bimodal throughout the process and achieving over 0.98 classification performance (AUC). Notably, a model trained on one or a few snapshots enables efficient on-the-fly computation of crystallinity. Lastly, we demonstrate how the optimal C-index fit evolves during various stages of crystallization, supporting the hypothesis that entropy dominates early nucleation, while symmetry gains relevance later. This workflow provides a data-driven strategy for OP selection and a metric to monitor structural transformations in large-scale polymer simulations.
GRAM-TDI: adaptive multimodal representation learning for drug target interaction prediction
Sep 26, 2025Drug target interaction (DTI) prediction is a cornerstone of computational drug discovery, enabling rational design, repurposing, and mechanistic insights. While deep learning has advanced DTI modeling, existing approaches primarily rely on SMILES protein pairs and fail to exploit the rich multimodal information available for small molecules and proteins. We introduce GRAMDTI, a pretraining framework that integrates multimodal molecular and protein inputs into unified representations. GRAMDTI extends volume based contrastive learning to four modalities, capturing higher-order semantic alignment beyond conventional pairwise approaches. To handle modality informativeness, we propose adaptive modality dropout, dynamically regulating each modality's contribution during pre-training. Additionally, IC50 activity measurements, when available, are incorporated as weak supervision to ground representations in biologically meaningful interaction strengths. Experiments on four publicly available datasets demonstrate that GRAMDTI consistently outperforms state of the art baselines. Our results highlight the benefits of higher order multimodal alignment, adaptive modality utilization, and auxiliary supervision for robust and generalizable DTI prediction.
Hybrid Quantum--Classical Machine Learning Potential with Variational Quantum Circuits
Aug 06, 2025



Quantum algorithms for simulating large and complex molecular systems are still in their infancy, and surpassing state-of-the-art classical techniques remains an ever-receding goal post. A promising avenue of inquiry in the meanwhile is to seek practical advantages through hybrid quantum-classical algorithms, which combine conventional neural networks with variational quantum circuits (VQCs) running on today's noisy intermediate-scale quantum (NISQ) hardware. Such hybrids are well suited to NISQ hardware. The classical processor performs the bulk of the computation, while the quantum processor executes targeted sub-tasks that supply additional non-linearity and expressivity. Here, we benchmark a purely classical E(3)-equivariant message-passing machine learning potential (MLP) against a hybrid quantum-classical MLP for predicting density functional theory (DFT) properties of liquid silicon. In our hybrid architecture, every readout in the message-passing layers is replaced by a VQC. Molecular dynamics simulations driven by the HQC-MLP reveal that an accurate reproduction of high-temperature structural and thermodynamic properties is achieved with VQCs. These findings demonstrate a concrete scenario in which NISQ-compatible HQC algorithm could deliver a measurable benefit over the best available classical alternative, suggesting a viable pathway toward near-term quantum advantage in materials modeling.
Iterative Pretraining Framework for Interatomic Potentials
Jul 27, 2025Machine learning interatomic potentials (MLIPs) enable efficient molecular dynamics (MD) simulations with ab initio accuracy and have been applied across various domains in physical science. However, their performance often relies on large-scale labeled training data. While existing pretraining strategies can improve model performance, they often suffer from a mismatch between the objectives of pretraining and downstream tasks or rely on extensive labeled datasets and increasingly complex architectures to achieve broad generalization. To address these challenges, we propose Iterative Pretraining for Interatomic Potentials (IPIP), a framework designed to iteratively improve the predictive performance of MLIP models. IPIP incorporates a forgetting mechanism to prevent iterative training from converging to suboptimal local minima. Unlike general-purpose foundation models, which frequently underperform on specialized tasks due to a trade-off between generality and system-specific accuracy, IPIP achieves higher accuracy and efficiency using lightweight architectures. Compared to general-purpose force fields, this approach achieves over 80% reduction in prediction error and up to 4x speedup in the challenging Mo-S-O system, enabling fast and accurate simulations.