 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSimilarity-aware Positive Instance Sampling for Graph Contrastive Pre-training
Jun 23, 2022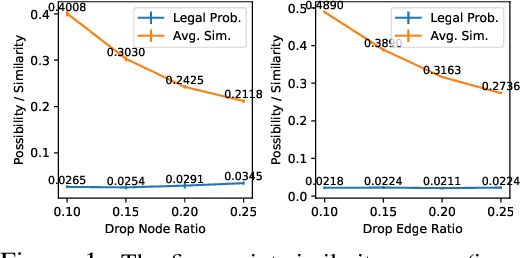
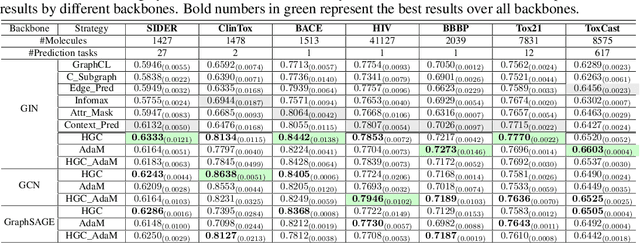
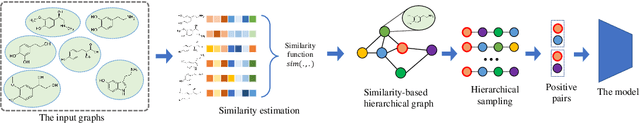
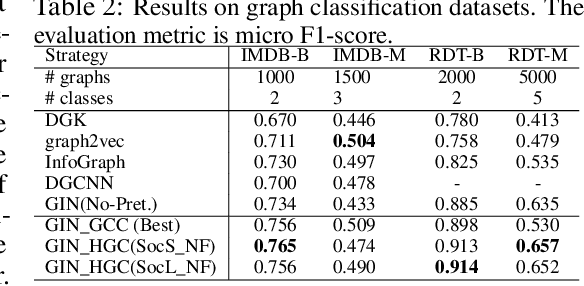
Graph instance contrastive learning has been proved as an effective task for Graph Neural Network (GNN) pre-training. However, one key issue may seriously impede the representative power in existing works: Positive instances created by current methods often miss crucial information of graphs or even yield illegal instances (such as non-chemically-aware graphs in molecular generation). To remedy this issue, we propose to select positive graph instances directly from existing graphs in the training set, which ultimately maintains the legality and similarity to the target graphs. Our selection is based on certain domain-specific pair-wise similarity measurements as well as sampling from a hierarchical graph encoding similarity relations among graphs. Besides, we develop an adaptive node-level pre-training method to dynamically mask nodes to distribute them evenly in the graph. We conduct extensive experiments on $13$ graph classification and node classification benchmark datasets from various domains. The results demonstrate that the GNN models pre-trained by our strategies can outperform those trained-from-scratch models as well as the variants obtained by existing methods.
Semi-Supervised Hierarchical Graph Classification
Jun 11, 2022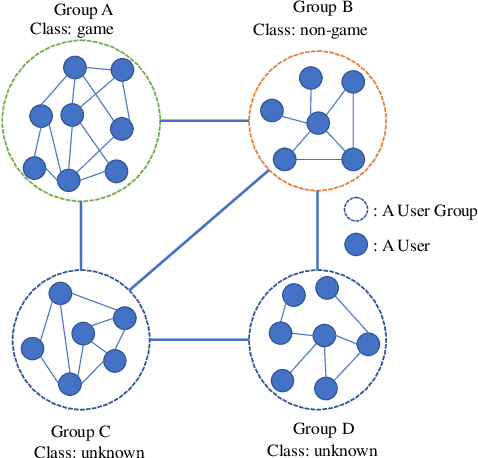
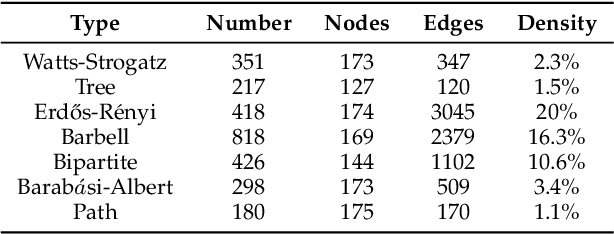
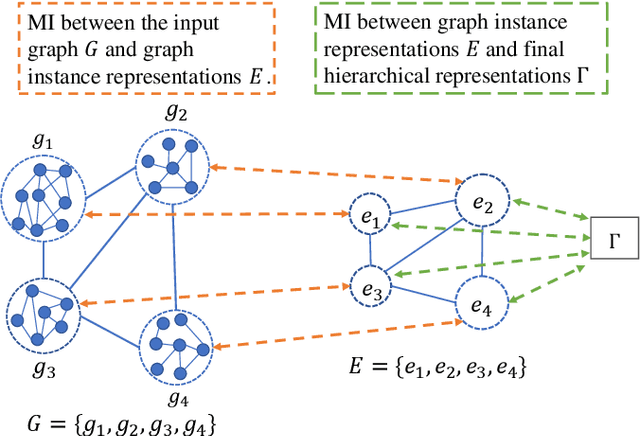
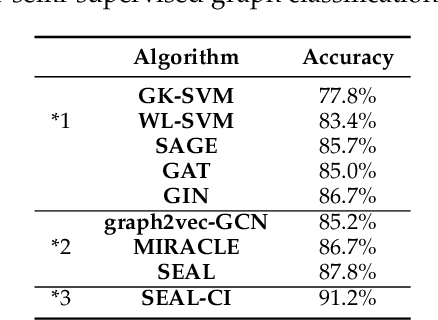
Node classification and graph classification are two graph learning problems that predict the class label of a node and the class label of a graph respectively. A node of a graph usually represents a real-world entity, e.g., a user in a social network, or a document in a document citation network. In this work, we consider a more challenging but practically useful setting, in which a node itself is a graph instance. This leads to a hierarchical graph perspective which arises in many domains such as social network, biological network and document collection. We study the node classification problem in the hierarchical graph where a 'node' is a graph instance. As labels are usually limited, we design a novel semi-supervised solution named SEAL-CI. SEAL-CI adopts an iterative framework that takes turns to update two modules, one working at the graph instance level and the other at the hierarchical graph level. To enforce a consistency among different levels of hierarchical graph, we propose the Hierarchical Graph Mutual Information (HGMI) and further present a way to compute HGMI with theoretical guarantee. We demonstrate the effectiveness of this hierarchical graph modeling and the proposed SEAL-CI method on text and social network data.
Towards Diverse and Natural Scene-aware 3D Human Motion Synthesis
May 25, 2022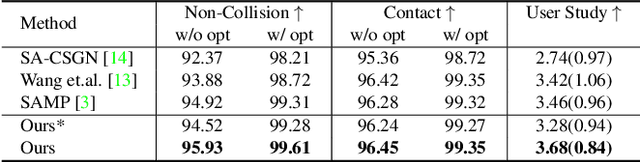

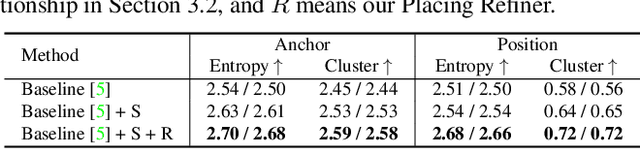
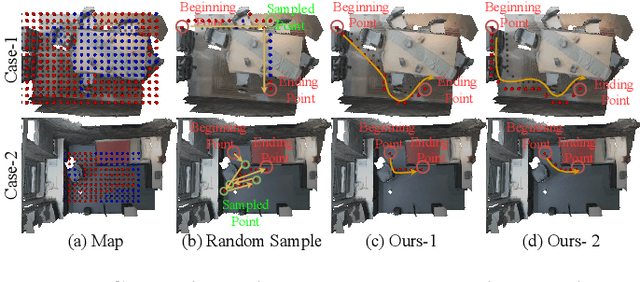
The ability to synthesize long-term human motion sequences in real-world scenes can facilitate numerous applications. Previous approaches for scene-aware motion synthesis are constrained by pre-defined target objects or positions and thus limit the diversity of human-scene interactions for synthesized motions. In this paper, we focus on the problem of synthesizing diverse scene-aware human motions under the guidance of target action sequences. To achieve this, we first decompose the diversity of scene-aware human motions into three aspects, namely interaction diversity (e.g. sitting on different objects with different poses in the given scenes), path diversity (e.g. moving to the target locations following different paths), and the motion diversity (e.g. having various body movements during moving). Based on this factorized scheme, a hierarchical framework is proposed, with each sub-module responsible for modeling one aspect. We assess the effectiveness of our framework on two challenging datasets for scene-aware human motion synthesis. The experiment results show that the proposed framework remarkably outperforms previous methods in terms of diversity and naturalness.
A Survey of Trustworthy Graph Learning: Reliability, Explainability, and Privacy Protection
May 23, 2022

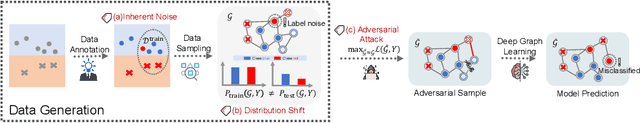
Deep graph learning has achieved remarkable progresses in both business and scientific areas ranging from finance and e-commerce, to drug and advanced material discovery. Despite these progresses, how to ensure various deep graph learning algorithms behave in a socially responsible manner and meet regulatory compliance requirements becomes an emerging problem, especially in risk-sensitive domains. Trustworthy graph learning (TwGL) aims to solve the above problems from a technical viewpoint. In contrast to conventional graph learning research which mainly cares about model performance, TwGL considers various reliability and safety aspects of the graph learning framework including but not limited to robustness, explainability, and privacy. In this survey, we provide a comprehensive review of recent leading approaches in the TwGL field from three dimensions, namely, reliability, explainability, and privacy protection. We give a general categorization for existing work and review typical work for each category. To give further insights for TwGL research, we provide a unified view to inspect previous works and build the connection between them. We also point out some important open problems remaining to be solved in the future developments of TwGL.
Hypergraph Convolutional Networks via Equivalency between Hypergraphs and Undirected Graphs
Apr 06, 2022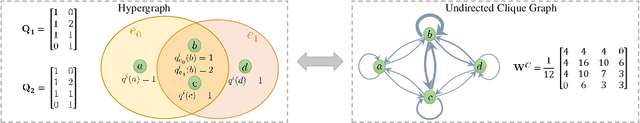
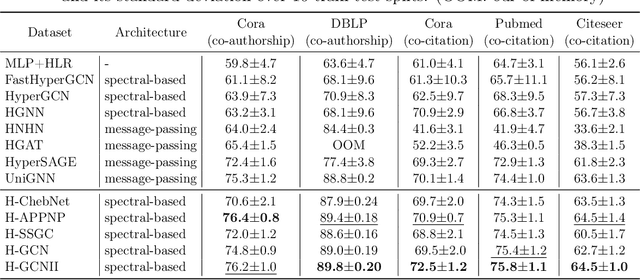
As a powerful tool for modeling complex relationships, hypergraphs are gaining popularity from the graph learning community. However, commonly used frameworks in deep hypergraph learning focus on hypergraphs with \textit{edge-independent vertex weights}(EIVWs), without considering hypergraphs with \textit{edge-dependent vertex weights} (EDVWs) that have more modeling power. To compensate for this, in this paper, we present General Hypergraph Spectral Convolution(GHSC), a general learning framework that not only can handle EDVW and EIVW hypergraphs, but more importantly, enables theoretically explicitly utilizing the existing powerful Graph Convolutional Neural Networks (GCNNs) such that largely ease the design of Hypergraph Neural Networks. In this framework, the graph Laplacian of the given undirected GCNNs is replaced with a unified hypergraph Laplacian that incorporates vertex weight information from a random walk perspective by equating our defined generalized hypergraphs with simple undirected graphs. Extensive experiments from various domains including social network analysis, visual objective classification, protein learning demonstrate that the proposed framework can achieve state-of-the-art performance.
Fine-Tuning Graph Neural Networks via Graph Topology induced Optimal Transport
Mar 20, 2022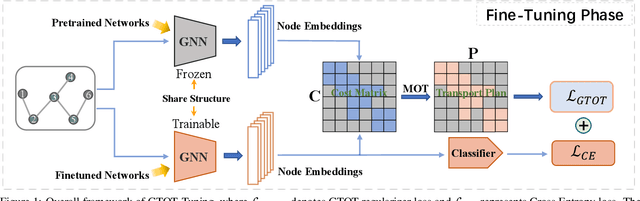
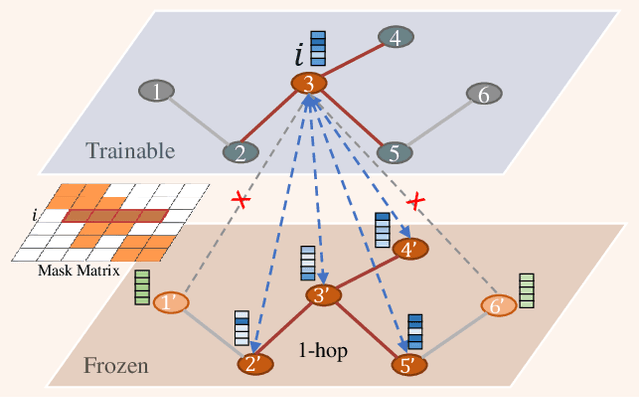
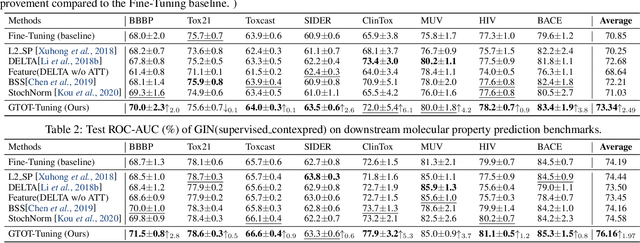
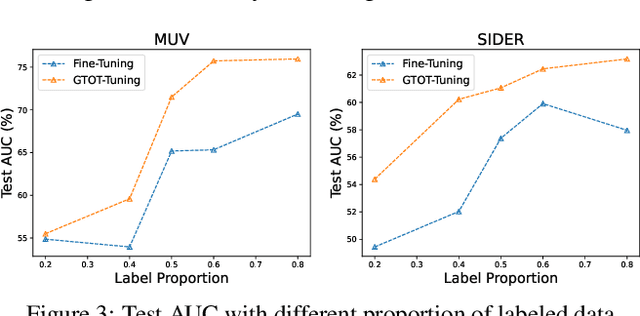
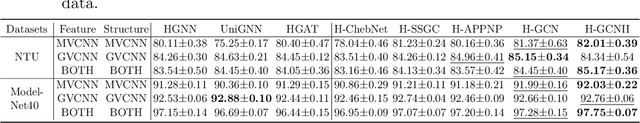
Recently, the pretrain-finetuning paradigm has attracted tons of attention in graph learning community due to its power of alleviating the lack of labels problem in many real-world applications. Current studies use existing techniques, such as weight constraint, representation constraint, which are derived from images or text data, to transfer the invariant knowledge from the pre-train stage to fine-tuning stage. However, these methods failed to preserve invariances from graph structure and Graph Neural Network (GNN) style models. In this paper, we present a novel optimal transport-based fine-tuning framework called GTOT-Tuning, namely, Graph Topology induced Optimal Transport fine-Tuning, for GNN style backbones. GTOT-Tuning is required to utilize the property of graph data to enhance the preservation of representation produced by fine-tuned networks. Toward this goal, we formulate graph local knowledge transfer as an Optimal Transport (OT) problem with a structural prior and construct the GTOT regularizer to constrain the fine-tuned model behaviors. By using the adjacency relationship amongst nodes, the GTOT regularizer achieves node-level optimal transport procedures and reduces redundant transport procedures, resulting in efficient knowledge transfer from the pre-trained models. We evaluate GTOT-Tuning on eight downstream tasks with various GNN backbones and demonstrate that it achieves state-of-the-art fine-tuning performance for GNNs.
Smoothing Matters: Momentum Transformer for Domain Adaptive Semantic Segmentation
Mar 15, 2022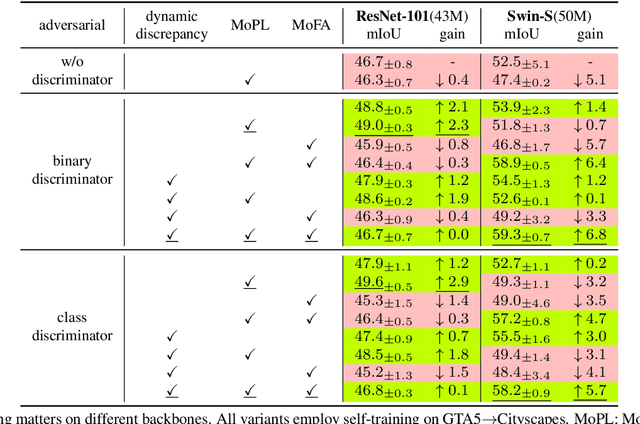
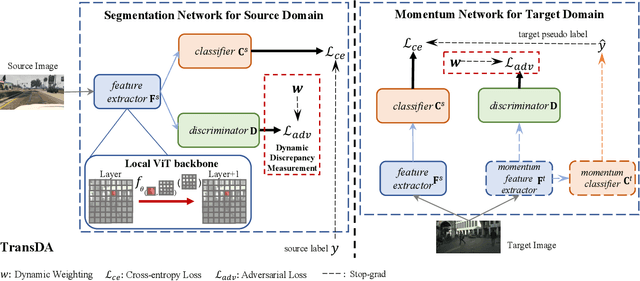

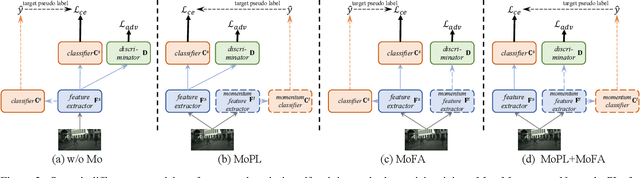
After the great success of Vision Transformer variants (ViTs) in computer vision, it has also demonstrated great potential in domain adaptive semantic segmentation. Unfortunately, straightforwardly applying local ViTs in domain adaptive semantic segmentation does not bring in expected improvement. We find that the pitfall of local ViTs is due to the severe high-frequency components generated during both the pseudo-label construction and features alignment for target domains. These high-frequency components make the training of local ViTs very unsmooth and hurt their transferability. In this paper, we introduce a low-pass filtering mechanism, momentum network, to smooth the learning dynamics of target domain features and pseudo labels. Furthermore, we propose a dynamic of discrepancy measurement to align the distributions in the source and target domains via dynamic weights to evaluate the importance of the samples. After tackling the above issues, extensive experiments on sim2real benchmarks show that the proposed method outperforms the state-of-the-art methods. Our codes are available at https://github.com/alpc91/TransDA
Equivariant Graph Mechanics Networks with Constraints
Mar 12, 2022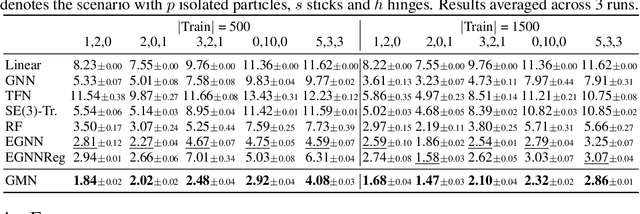
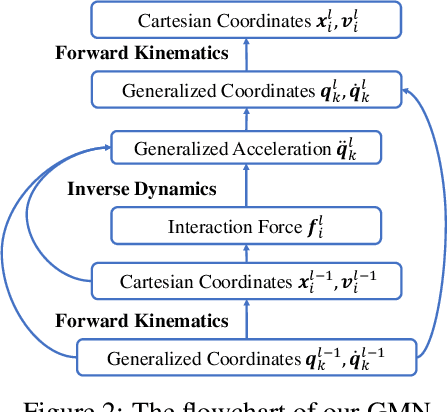
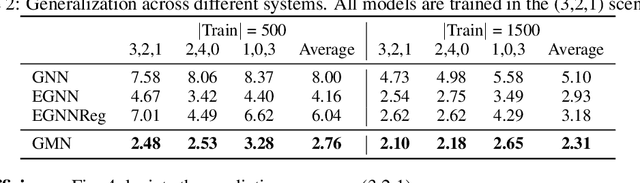
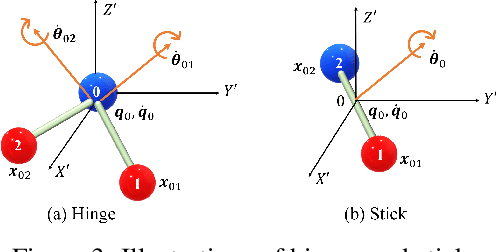
Learning to reason about relations and dynamics over multiple interacting objects is a challenging topic in machine learning. The challenges mainly stem from that the interacting systems are exponentially-compositional, symmetrical, and commonly geometrically-constrained. Current methods, particularly the ones based on equivariant Graph Neural Networks (GNNs), have targeted on the first two challenges but remain immature for constrained systems. In this paper, we propose Graph Mechanics Network (GMN) which is combinatorially efficient, equivariant and constraint-aware. The core of GMN is that it represents, by generalized coordinates, the forward kinematics information (positions and velocities) of a structural object. In this manner, the geometrical constraints are implicitly and naturally encoded in the forward kinematics. Moreover, to allow equivariant message passing in GMN, we have developed a general form of orthogonality-equivariant functions, given that the dynamics of constrained systems are more complicated than the unconstrained counterparts. Theoretically, the proposed equivariant formulation is proved to be universally expressive under certain conditions. Extensive experiments support the advantages of GMN compared to the state-of-the-art GNNs in terms of prediction accuracy, constraint satisfaction and data efficiency on the simulated systems consisting of particles, sticks and hinges, as well as two real-world datasets for molecular dynamics prediction and human motion capture.
Geometrically Equivariant Graph Neural Networks: A Survey
Feb 22, 2022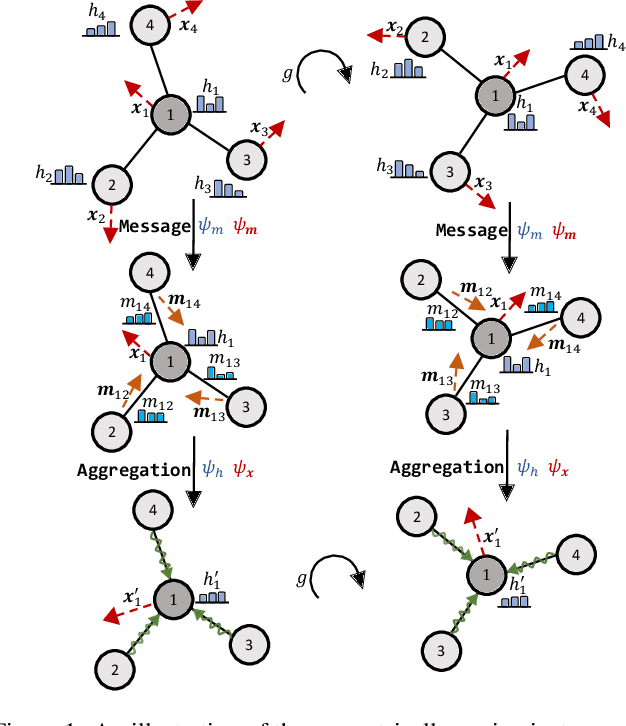
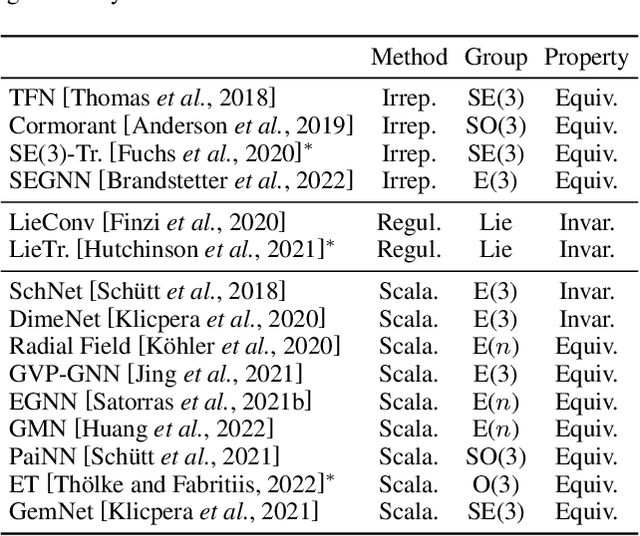
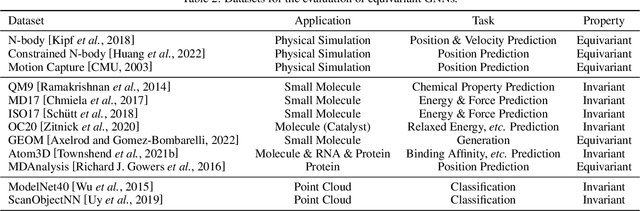
Many scientific problems require to process data in the form of geometric graphs. Unlike generic graph data, geometric graphs exhibit symmetries of translations, rotations, and/or reflections. Researchers have leveraged such inductive bias and developed geometrically equivariant Graph Neural Networks (GNNs) to better characterize the geometry and topology of geometric graphs. Despite fruitful achievements, it still lacks a survey to depict how equivariant GNNs are progressed, which in turn hinders the further development of equivariant GNNs. To this end, based on the necessary but concise mathematical preliminaries, we analyze and classify existing methods into three groups regarding how the message passing and aggregation in GNNs are represented. We also summarize the benchmarks as well as the related datasets to facilitate later researches for methodology development and experimental evaluation. The prospect for future potential directions is also provided.
Equivariant Graph Hierarchy-Based Neural Networks
Feb 22, 2022

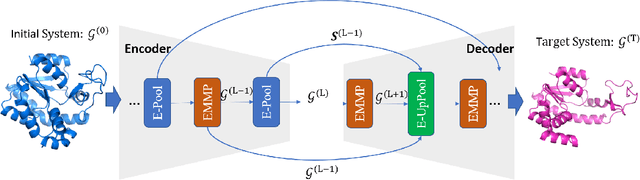
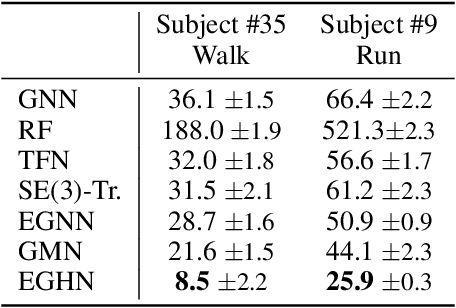
Equivariant Graph neural Networks (EGNs) are powerful in characterizing the dynamics of multi-body physical systems. Existing EGNs conduct flat message passing, which, yet, is unable to capture the spatial/dynamical hierarchy for complex systems particularly, limiting substructure discovery and global information fusion. In this paper, we propose Equivariant Hierarchy-based Graph Networks (EGHNs) which consist of the three key components: generalized Equivariant Matrix Message Passing (EMMP) , E-Pool and E-UpPool. In particular, EMMP is able to improve the expressivity of conventional equivariant message passing, E-Pool assigns the quantities of the low-level nodes into high-level clusters, while E-UpPool leverages the high-level information to update the dynamics of the low-level nodes. As their names imply, both E-Pool and E-UpPool are guaranteed to be equivariant to meet physic symmetry. Considerable experimental evaluations verify the effectiveness of our EGHN on several applications including multi-object dynamics simulation, motion capture, and protein dynamics modeling.