 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeLifespan Pancreas Morphology for Control vs Type 2 Diabetes using AI on Largescale Clinical Imaging
Aug 20, 2025Purpose: Understanding how the pancreas changes is critical for detecting deviations in type 2 diabetes and other pancreatic disease. We measure pancreas size and shape using morphological measurements from ages 0 to 90. Our goals are to 1) identify reliable clinical imaging modalities for AI-based pancreas measurement, 2) establish normative morphological aging trends, and 3) detect potential deviations in type 2 diabetes. Approach: We analyzed a clinically acquired dataset of 2533 patients imaged with abdominal CT or MRI. We resampled the scans to 3mm isotropic resolution, segmented the pancreas using automated methods, and extracted 13 morphological pancreas features across the lifespan. First, we assessed CT and MRI measurements to determine which modalities provide consistent lifespan trends. Second, we characterized distributions of normative morphological patterns stratified by age group and sex. Third, we used GAMLSS regression to model pancreas morphology trends in 1350 patients matched for age, sex, and type 2 diabetes status to identify any deviations from normative aging associated with type 2 diabetes. Results: When adjusting for confounders, the aging trends for 10 of 13 morphological features were significantly different between patients with type 2 diabetes and non-diabetic controls (p < 0.05 after multiple comparisons corrections). Additionally, MRI appeared to yield different pancreas measurements than CT using our AI-based method. Conclusions: We provide lifespan trends demonstrating that the size and shape of the pancreas is altered in type 2 diabetes using 675 control patients and 675 diabetes patients. Moreover, our findings reinforce that the pancreas is smaller in type 2 diabetes. Additionally, we contribute a reference of lifespan pancreas morphology from a large cohort of non-diabetic control patients in a clinical setting.
Cohort-Aware Agents for Individualized Lung Cancer Risk Prediction Using a Retrieval-Augmented Model Selection Framework
Aug 20, 2025



Accurate lung cancer risk prediction remains challenging due to substantial variability across patient populations and clinical settings -- no single model performs best for all cohorts. To address this, we propose a personalized lung cancer risk prediction agent that dynamically selects the most appropriate model for each patient by combining cohort-specific knowledge with modern retrieval and reasoning techniques. Given a patient's CT scan and structured metadata -- including demographic, clinical, and nodule-level features -- the agent first performs cohort retrieval using FAISS-based similarity search across nine diverse real-world cohorts to identify the most relevant patient population from a multi-institutional database. Second, a Large Language Model (LLM) is prompted with the retrieved cohort and its associated performance metrics to recommend the optimal prediction algorithm from a pool of eight representative models, including classical linear risk models (e.g., Mayo, Brock), temporally-aware models (e.g., TDVIT, DLSTM), and multi-modal computer vision-based approaches (e.g., Liao, Sybil, DLS, DLI). This two-stage agent pipeline -- retrieval via FAISS and reasoning via LLM -- enables dynamic, cohort-aware risk prediction personalized to each patient's profile. Building on this architecture, the agent supports flexible and cohort-driven model selection across diverse clinical populations, offering a practical path toward individualized risk assessment in real-world lung cancer screening.
Multipath cycleGAN for harmonization of paired and unpaired low-dose lung computed tomography reconstruction kernels
May 28, 2025Reconstruction kernels in computed tomography (CT) affect spatial resolution and noise characteristics, introducing systematic variability in quantitative imaging measurements such as emphysema quantification. Choosing an appropriate kernel is therefore essential for consistent quantitative analysis. We propose a multipath cycleGAN model for CT kernel harmonization, trained on a mixture of paired and unpaired data from a low-dose lung cancer screening cohort. The model features domain-specific encoders and decoders with a shared latent space and uses discriminators tailored for each domain.We train the model on 42 kernel combinations using 100 scans each from seven representative kernels in the National Lung Screening Trial (NLST) dataset. To evaluate performance, 240 scans from each kernel are harmonized to a reference soft kernel, and emphysema is quantified before and after harmonization. A general linear model assesses the impact of age, sex, smoking status, and kernel on emphysema. We also evaluate harmonization from soft kernels to a reference hard kernel. To assess anatomical consistency, we compare segmentations of lung vessels, muscle, and subcutaneous adipose tissue generated by TotalSegmentator between harmonized and original images. Our model is benchmarked against traditional and switchable cycleGANs. For paired kernels, our approach reduces bias in emphysema scores, as seen in Bland-Altman plots (p<0.05). For unpaired kernels, harmonization eliminates confounding differences in emphysema (p>0.05). High Dice scores confirm preservation of muscle and fat anatomy, while lung vessel overlap remains reasonable. Overall, our shared latent space multipath cycleGAN enables robust harmonization across paired and unpaired CT kernels, improving emphysema quantification and preserving anatomical fidelity.
Rep3D: Re-parameterize Large 3D Kernels with Low-Rank Receptive Modeling for Medical Imaging
May 26, 2025In contrast to vision transformers, which model long-range dependencies through global self-attention, large kernel convolutions provide a more efficient and scalable alternative, particularly in high-resolution 3D volumetric settings. However, naively increasing kernel size often leads to optimization instability and degradation in performance. Motivated by the spatial bias observed in effective receptive fields (ERFs), we hypothesize that different kernel elements converge at variable rates during training. To support this, we derive a theoretical connection between element-wise gradients and first-order optimization, showing that structurally re-parameterized convolution blocks inherently induce spatially varying learning rates. Building on this insight, we introduce Rep3D, a 3D convolutional framework that incorporates a learnable spatial prior into large kernel training. A lightweight two-stage modulation network generates a receptive-biased scaling mask, adaptively re-weighting kernel updates and enabling local-to-global convergence behavior. Rep3D adopts a plain encoder design with large depthwise convolutions, avoiding the architectural complexity of multi-branch compositions. We evaluate Rep3D on five challenging 3D segmentation benchmarks and demonstrate consistent improvements over state-of-the-art baselines, including transformer-based and fixed-prior re-parameterization methods. By unifying spatial inductive bias with optimization-aware learning, Rep3D offers an interpretable, and scalable solution for 3D medical image analysis. The source code is publicly available at https://github.com/leeh43/Rep3D.
KPIs 2024 Challenge: Advancing Glomerular Segmentation from Patch- to Slide-Level
Feb 11, 2025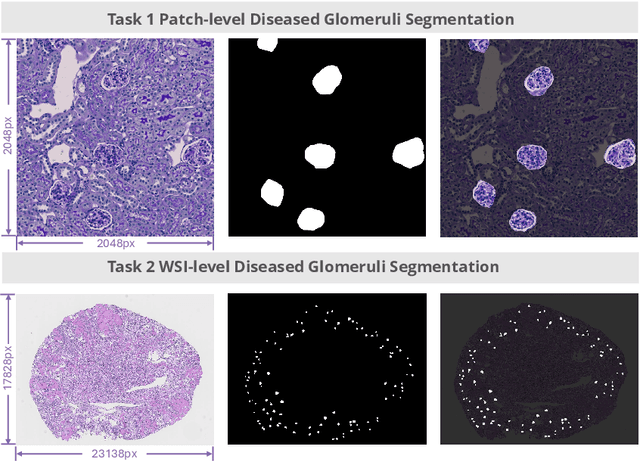
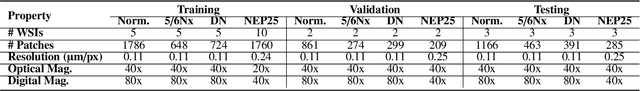
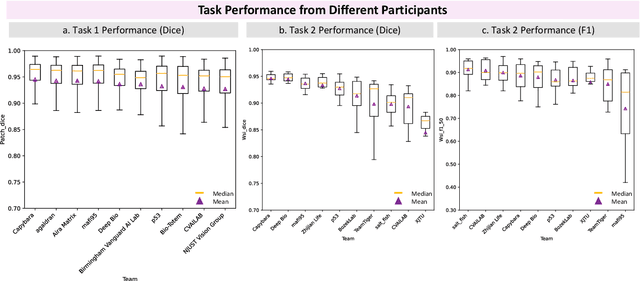

Chronic kidney disease (CKD) is a major global health issue, affecting over 10% of the population and causing significant mortality. While kidney biopsy remains the gold standard for CKD diagnosis and treatment, the lack of comprehensive benchmarks for kidney pathology segmentation hinders progress in the field. To address this, we organized the Kidney Pathology Image Segmentation (KPIs) Challenge, introducing a dataset that incorporates preclinical rodent models of CKD with over 10,000 annotated glomeruli from 60+ Periodic Acid Schiff (PAS)-stained whole slide images. The challenge includes two tasks, patch-level segmentation and whole slide image segmentation and detection, evaluated using the Dice Similarity Coefficient (DSC) and F1-score. By encouraging innovative segmentation methods that adapt to diverse CKD models and tissue conditions, the KPIs Challenge aims to advance kidney pathology analysis, establish new benchmarks, and enable precise, large-scale quantification for disease research and diagnosis.
Investigating the impact of kernel harmonization and deformable registration on inspiratory and expiratory chest CT images for people with COPD
Feb 07, 2025



Paired inspiratory-expiratory CT scans enable the quantification of gas trapping due to small airway disease and emphysema by analyzing lung tissue motion in COPD patients. Deformable image registration of these scans assesses regional lung volumetric changes. However, variations in reconstruction kernels between paired scans introduce errors in quantitative analysis. This work proposes a two-stage pipeline to harmonize reconstruction kernels and perform deformable image registration using data acquired from the COPDGene study. We use a cycle generative adversarial network (GAN) to harmonize inspiratory scans reconstructed with a hard kernel (BONE) to match expiratory scans reconstructed with a soft kernel (STANDARD). We then deformably register the expiratory scans to inspiratory scans. We validate harmonization by measuring emphysema using a publicly available segmentation algorithm before and after harmonization. Results show harmonization significantly reduces emphysema measurement inconsistencies, decreasing median emphysema scores from 10.479% to 3.039%, with a reference median score of 1.305% from the STANDARD kernel as the target. Registration accuracy is evaluated via Dice overlap between emphysema regions on inspiratory, expiratory, and deformed images. The Dice coefficient between inspiratory emphysema masks and deformably registered emphysema masks increases significantly across registration stages (p<0.001). Additionally, we demonstrate that deformable registration is robust to kernel variations.
Pitfalls of defacing whole-head MRI: re-identification risk with diffusion models and compromised research potential
Jan 31, 2025Defacing is often applied to head magnetic resonance image (MRI) datasets prior to public release to address privacy concerns. The alteration of facial and nearby voxels has provoked discussions about the true capability of these techniques to ensure privacy as well as their impact on downstream tasks. With advancements in deep generative models, the extent to which defacing can protect privacy is uncertain. Additionally, while the altered voxels are known to contain valuable anatomical information, their potential to support research beyond the anatomical regions directly affected by defacing remains uncertain. To evaluate these considerations, we develop a refacing pipeline that recovers faces in defaced head MRIs using cascaded diffusion probabilistic models (DPMs). The DPMs are trained on images from 180 subjects and tested on images from 484 unseen subjects, 469 of whom are from a different dataset. To assess whether the altered voxels in defacing contain universally useful information, we also predict computed tomography (CT)-derived skeletal muscle radiodensity from facial voxels in both defaced and original MRIs. The results show that DPMs can generate high-fidelity faces that resemble the original faces from defaced images, with surface distances to the original faces significantly smaller than those of a population average face (p < 0.05). This performance also generalizes well to previously unseen datasets. For skeletal muscle radiodensity predictions, using defaced images results in significantly weaker Spearman's rank correlation coefficients compared to using original images (p < 10-4). For shin muscle, the correlation is statistically significant (p < 0.05) when using original images but not statistically significant (p > 0.05) when any defacing method is applied, suggesting that defacing might not only fail to protect privacy but also eliminate valuable information.
Post-Training Quantization for 3D Medical Image Segmentation: A Practical Study on Real Inference Engines
Jan 28, 2025Quantizing deep neural networks ,reducing the precision (bit-width) of their computations, can remarkably decrease memory usage and accelerate processing, making these models more suitable for large-scale medical imaging applications with limited computational resources. However, many existing methods studied "fake quantization", which simulates lower precision operations during inference, but does not actually reduce model size or improve real-world inference speed. Moreover, the potential of deploying real 3D low-bit quantization on modern GPUs is still unexplored. In this study, we introduce a real post-training quantization (PTQ) framework that successfully implements true 8-bit quantization on state-of-the-art (SOTA) 3D medical segmentation models, i.e., U-Net, SegResNet, SwinUNETR, nnU-Net, UNesT, TransUNet, ST-UNet,and VISTA3D. Our approach involves two main steps. First, we use TensorRT to perform fake quantization for both weights and activations with unlabeled calibration dataset. Second, we convert this fake quantization into real quantization via TensorRT engine on real GPUs, resulting in real-world reductions in model size and inference latency. Extensive experiments demonstrate that our framework effectively performs 8-bit quantization on GPUs without sacrificing model performance. This advancement enables the deployment of efficient deep learning models in medical imaging applications where computational resources are constrained. The code and models have been released, including U-Net, TransUNet pretrained on the BTCV dataset for abdominal (13-label) segmentation, UNesT pretrained on the Whole Brain Dataset for whole brain (133-label) segmentation, and nnU-Net, SegResNet, SwinUNETR and VISTA3D pretrained on TotalSegmentator V2 for full body (104-label) segmentation. https://github.com/hrlblab/PTQ.
MICCAI-CDMRI 2023 QuantConn Challenge Findings on Achieving Robust Quantitative Connectivity through Harmonized Preprocessing of Diffusion MRI
Nov 14, 2024



White matter alterations are increasingly implicated in neurological diseases and their progression. International-scale studies use diffusion-weighted magnetic resonance imaging (DW-MRI) to qualitatively identify changes in white matter microstructure and connectivity. Yet, quantitative analysis of DW-MRI data is hindered by inconsistencies stemming from varying acquisition protocols. There is a pressing need to harmonize the preprocessing of DW-MRI datasets to ensure the derivation of robust quantitative diffusion metrics across acquisitions. In the MICCAI-CDMRI 2023 QuantConn challenge, participants were provided raw data from the same individuals collected on the same scanner but with two different acquisitions and tasked with preprocessing the DW-MRI to minimize acquisition differences while retaining biological variation. Submissions are evaluated on the reproducibility and comparability of cross-acquisition bundle-wise microstructure measures, bundle shape features, and connectomics. The key innovations of the QuantConn challenge are that (1) we assess bundles and tractography in the context of harmonization for the first time, (2) we assess connectomics in the context of harmonization for the first time, and (3) we have 10x additional subjects over prior harmonization challenge, MUSHAC and 100x over SuperMUDI. We find that bundle surface area, fractional anisotropy, connectome assortativity, betweenness centrality, edge count, modularity, nodal strength, and participation coefficient measures are most biased by acquisition and that machine learning voxel-wise correction, RISH mapping, and NeSH methods effectively reduce these biases. In addition, microstructure measures AD, MD, RD, bundle length, connectome density, efficiency, and path length are least biased by these acquisition differences.
* Accepted for publication at the Journal of Machine Learning for Biomedical Imaging (MELBA) https://melba-journal.org/2024/019
Linear Spherical Sliced Optimal Transport: A Fast Metric for Comparing Spherical Data
Nov 09, 2024



Efficient comparison of spherical probability distributions becomes important in fields such as computer vision, geosciences, and medicine. Sliced optimal transport distances, such as spherical and stereographic spherical sliced Wasserstein distances, have recently been developed to address this need. These methods reduce the computational burden of optimal transport by slicing hyperspheres into one-dimensional projections, i.e., lines or circles. Concurrently, linear optimal transport has been proposed to embed distributions into \( L^2 \) spaces, where the \( L^2 \) distance approximates the optimal transport distance, thereby simplifying comparisons across multiple distributions. In this work, we introduce the Linear Spherical Sliced Optimal Transport (LSSOT) framework, which utilizes slicing to embed spherical distributions into \( L^2 \) spaces while preserving their intrinsic geometry, offering a computationally efficient metric for spherical probability measures. We establish the metricity of LSSOT and demonstrate its superior computational efficiency in applications such as cortical surface registration, 3D point cloud interpolation via gradient flow, and shape embedding. Our results demonstrate the significant computational benefits and high accuracy of LSSOT in these applications.