 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeRadOT-Eval: Auditable Structured-Evidence Transport for Radiology Report Evaluation
Jun 07, 2026Automatic evaluation is critical for high-stakes text generation, where errors often involve omitted findings, hallucinated content, polarity reversals, location changes, uncertainty mismatches, and temporal-comparison errors rather than low surface similarity alone. Radiology report generation provides a challenging test case because generated reports must preserve structured clinical evidence across sources. We present RadOT-Eval, an interpretable structured-evidence optimal transport framework for offline auditing of radiology report generation. RadOT-Eval decomposes reference and candidate reports into attribute-structured clinical evidence units, aligns corresponding evidence using entropy-regularized optimal transport, and uses clinically meaningful side-channel discrepancies in a monotone risk model to predict error burden. All transport, feature, and readout choices are selected using the ReXVal dataset, and the frozen system is evaluated on the independent RadEvalX dataset. RadOT-Eval achieves Spearman correlations of 0.715, 0.548, and 0.399 with total, clinically significant, and clinically insignificant annotated error burden, respectively, yielding higher point estimates than standard evaluation metrics and the open-source large language model (LLM)-based evaluator GREEN-radllama2-7B. In a frozen auxiliary corruption-sensitivity stress test on ReXErr-v1, RadOT-Eval achieves 0.768 AUROC and a 0.990 corrupted-greater-than-clean paired win rate. These results show that structured evidence transport provides an auditable, rank-oriented evaluation tool for high-stakes generated clinical text under ReXVal-only model selection and frozen RadEvalX testing.
It's Not Always Sycophancy: Measuring LLM Conformity as a Function of Epistemic Uncertainty
May 26, 2026Large language models (LLMs) are known to abandon their initial stance to conform to user pushback. While prior research largely attributes this behavior to sycophancy learned during reinforcement learning from human feedback, we hypothesize that conformity is also driven by a model's epistemic uncertainty at inference time. In this paper, we introduce MUSE, a two-stage evaluation framework to disentangle the mechanisms driving LLM conformity. Specifically, MUSE maps a model's epistemic uncertainty in responding to a query against its likelihood to yield to user pushback in a subsequent turn. We demonstrate that the mechanisms driving conformity extend beyond sycophancy alone. Specifically, we characterize two distinct factors that jointly drive conformity: sycophantic conformity, where a model aligns with user pushback even with absolute certainty in its initial response, and uncertainty-driven conformity, where a model's likelihood for conformity increases alongside its uncertainty. Furthermore, we conduct ablation studies to demonstrate that both sycophantic conformity and uncertainty-driven conformity grow with 1) the LLM's perceived expertise of the user and 2) the plausibility of the user's suggestions. More broadly, MUSE informs more targeted intervention strategies by distinguishing alignment-induced sycophancy and training-corpora-driven uncertainty.
Vectors Are Not Neutral: Sensitive-Information Inference from Exported LLM Representations in Summarization
May 26, 2026Large language model (LLM) summarization systems may pass compact vector representations of private inputs to downstream retrieval, monitoring, audit, or analytic workflows. Even when source documents remain access-restricted, derived vectors may be handled under different access controls and still support sensitive-information inference, creating a residual information-disclosure risk. We study this issue in clinical discharge-summary generation as a high-stakes case study, using electronic health record (EHR)-recorded race as a controlled sensitive-label audit. We audit two artifacts that a system might retain or expose to downstream components: the final prompt-token hidden state and the mean-pooled prompt representation. Our results show that reducing recoverability of the case-study sensitive label from one exported artifact does not necessarily reduce recoverability from another. As a mitigation case study, we introduce SurfaceLoRA, an exported-vector-targeted parameter-efficient fine-tuning method that uses a gradient-reversal discriminator attached to a designated exported vector. Under a balanced five-way probing protocol, SurfaceLoRA reduces EHR-recorded race recoverability from the targeted final-token artifact toward chance while preserving summarization utility, yet recoverability remains substantially higher from untargeted pooled artifacts. These findings show that privacy auditing and mitigation should be performed on the exact vector artifact retained or exposed to downstream components.
DUET: Dual-Paradigm Adaptive Expert Triage with Single-cell Inductive Prior for Spatial Transcriptomics Prediction
May 13, 2026Inferring spatially resolved gene expression from histology images offers a cost-effective complement to spatial transcriptomics (ST). However, existing methods reduce this task to a simple morphology-to-expression mapping, where visual similarity does not guarantee molecular consistency. Meanwhile, single-cell data has amassed rich resources far surpassing the scale of ST data, yet it remains underexplored in vision-omics modeling. Furthermore, current approaches commit to a monolithic paradigm with bottlenecks, unable to balance expressive flexibility with biological fidelity. To bridge these gaps, we propose DUET, a novel dual-paradigm framework that synergizes parametric prediction and memory-based retrieval under cellular inductive priors. DUET implements a parallel regression-retrieval paradigm, adaptively reconciling the outputs of its complementary pathways. To mitigate aleatoric vision ambiguity, we incorporate large-scale single-cell references to impose molecular states as biological constraints for faithful learning. Building upon structural refinement, we further design a lightweight adapter to dynamically assign branch preference across spatial contexts to achieve optimal performance. Extensive experiments on three public datasets across varied gene scales demonstrate that DUET achieves SOTA performance, with consistent gains contributed by each proposed component. Code is available at https://github.com/Junchao-Zhu/DUET
Stop Listening to Me! How Multi-turn Conversations Can Degrade Diagnostic Reasoning
Mar 12, 2026Patients and clinicians are increasingly using chatbots powered by large language models (LLMs) for healthcare inquiries. While state-of-the-art LLMs exhibit high performance on static diagnostic reasoning benchmarks, their efficacy across multi-turn conversations, which better reflect real-world usage, has been understudied. In this paper, we evaluate 17 LLMs across three clinical datasets to investigate how partitioning the decision-space into multiple simpler turns of conversation influences their diagnostic reasoning. Specifically, we develop a "stick-or-switch" evaluation framework to measure model conviction (i.e., defending a correct diagnosis or safe abstention against incorrect suggestions) and flexibility (i.e., recognizing a correct suggestion when it is introduced) across conversations. Our experiments reveal the conversation tax, where multi-turn interactions consistently degrade performance when compared to single-shot baselines. Notably, models frequently abandon initial correct diagnoses and safe abstentions to align with incorrect user suggestions. Additionally, several models exhibit blind switching, failing to distinguish between signal and incorrect suggestions.
Learning When to Sample: Confidence-Aware Self-Consistency for Efficient LLM Chain-of-Thought Reasoning
Mar 09, 2026Large language models (LLMs) achieve strong reasoning performance through chain-of-thought (CoT) reasoning, yet often generate unnecessarily long reasoning paths that incur high inference cost. Recent self-consistency-based approaches further improve accuracy but require sampling and aggregating multiple reasoning trajectories, leading to substantial additional computational overhead. This paper introduces a confidence-aware decision framework that analyzes a single completed reasoning trajectory to adaptively select between single-path and multi-path reasoning. The framework is trained using sentence-level numeric and linguistic features extracted from intermediate reasoning states in the MedQA dataset and generalizes effectively to MathQA, MedMCQA, and MMLU without additional fine-tuning. Experimental results show that the proposed method maintains accuracy comparable to multi-path baselines while using up to 80\% fewer tokens. These findings demonstrate that reasoning trajectories contain rich signals for uncertainty estimation, enabling a simple, transferable mechanism to balance accuracy and efficiency in LLM reasoning.
AdaFuse: Adaptive Multimodal Fusion for Lung Cancer Risk Prediction via Reinforcement Learning
Jan 30, 2026Multimodal fusion has emerged as a promising paradigm for disease diagnosis and prognosis, integrating complementary information from heterogeneous data sources such as medical images, clinical records, and radiology reports. However, existing fusion methods process all available modalities through the network, either treating them equally or learning to assign different contribution weights, leaving a fundamental question unaddressed: for a given patient, should certain modalities be used at all? We present AdaFuse, an adaptive multimodal fusion framework that leverages reinforcement learning (RL) to learn patient-specific modality selection and fusion strategies for lung cancer risk prediction. AdaFuse formulates multimodal fusion as a sequential decision process, where the policy network iteratively decides whether to incorporate an additional modality or proceed to prediction based on the information already acquired. This sequential formulation enables the model to condition each selection on previously observed modalities and terminate early when sufficient information is available, rather than committing to a fixed subset upfront. We evaluate AdaFuse on the National Lung Screening Trial (NLST) dataset. Experimental results demonstrate that AdaFuse achieves the highest AUC (0.762) compared to the best single-modality baseline (0.732), the best fixed fusion strategy (0.759), and adaptive baselines including DynMM (0.754) and MoE (0.742), while using fewer FLOPs than all triple-modality methods. Our work demonstrates the potential of reinforcement learning for personalized multimodal fusion in medical imaging, representing a shift from uniform fusion strategies toward adaptive diagnostic pipelines that learn when to consult additional modalities and when existing information suffices for accurate prediction.
SCR2-ST: Combine Single Cell with Spatial Transcriptomics for Efficient Active Sampling via Reinforcement Learning
Dec 15, 2025



Spatial transcriptomics (ST) is an emerging technology that enables researchers to investigate the molecular relationships underlying tissue morphology. However, acquiring ST data remains prohibitively expensive, and traditional fixed-grid sampling strategies lead to redundant measurements of morphologically similar or biologically uninformative regions, thus resulting in scarce data that constrain current methods. The well-established single-cell sequencing field, however, could provide rich biological data as an effective auxiliary source to mitigate this limitation. To bridge these gaps, we introduce SCR2-ST, a unified framework that leverages single-cell prior knowledge to guide efficient data acquisition and accurate expression prediction. SCR2-ST integrates a single-cell guided reinforcement learning-based (SCRL) active sampling and a hybrid regression-retrieval prediction network SCR2Net. SCRL combines single-cell foundation model embeddings with spatial density information to construct biologically grounded reward signals, enabling selective acquisition of informative tissue regions under constrained sequencing budgets. SCR2Net then leverages the actively sampled data through a hybrid architecture combining regression-based modeling with retrieval-augmented inference, where a majority cell-type filtering mechanism suppresses noisy matches and retrieved expression profiles serve as soft labels for auxiliary supervision. We evaluated SCR2-ST on three public ST datasets, demonstrating SOTA performance in both sampling efficiency and prediction accuracy, particularly under low-budget scenarios. Code is publicly available at: https://github.com/hrlblab/SCR2ST
How Close Are We? Limitations and Progress of AI Models in Banff Lesion Scoring
Oct 31, 2025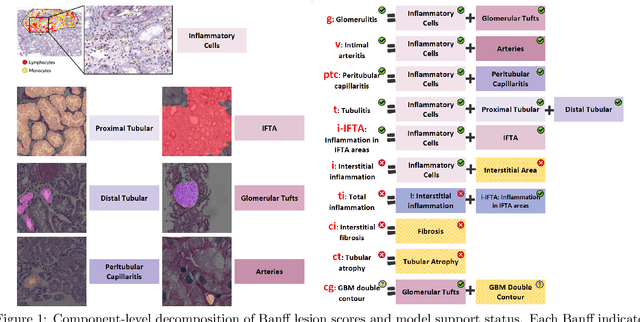
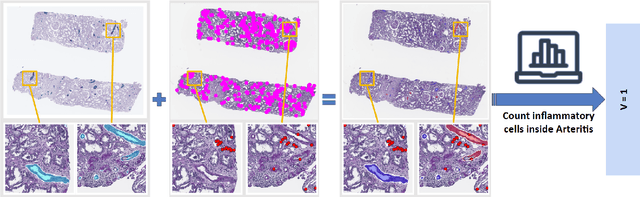
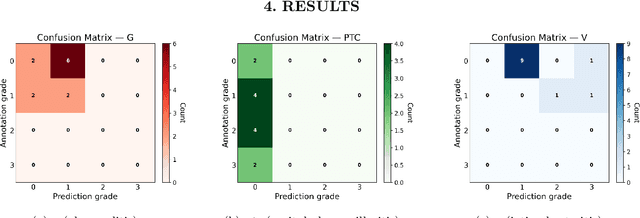
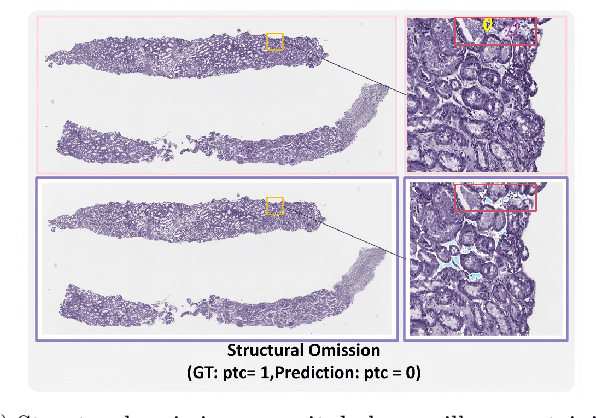
The Banff Classification provides the global standard for evaluating renal transplant biopsies, yet its semi-quantitative nature, complex criteria, and inter-observer variability present significant challenges for computational replication. In this study, we explore the feasibility of approximating Banff lesion scores using existing deep learning models through a modular, rule-based framework. We decompose each Banff indicator - such as glomerulitis (g), peritubular capillaritis (ptc), and intimal arteritis (v) - into its constituent structural and inflammatory components, and assess whether current segmentation and detection tools can support their computation. Model outputs are mapped to Banff scores using heuristic rules aligned with expert guidelines, and evaluated against expert-annotated ground truths. Our findings highlight both partial successes and critical failure modes, including structural omission, hallucination, and detection ambiguity. Even when final scores match expert annotations, inconsistencies in intermediate representations often undermine interpretability. These results reveal the limitations of current AI pipelines in replicating computational expert-level grading, and emphasize the importance of modular evaluation and computational Banff grading standard in guiding future model development for transplant pathology.
Cohort-Aware Agents for Individualized Lung Cancer Risk Prediction Using a Retrieval-Augmented Model Selection Framework
Aug 20, 2025



Accurate lung cancer risk prediction remains challenging due to substantial variability across patient populations and clinical settings -- no single model performs best for all cohorts. To address this, we propose a personalized lung cancer risk prediction agent that dynamically selects the most appropriate model for each patient by combining cohort-specific knowledge with modern retrieval and reasoning techniques. Given a patient's CT scan and structured metadata -- including demographic, clinical, and nodule-level features -- the agent first performs cohort retrieval using FAISS-based similarity search across nine diverse real-world cohorts to identify the most relevant patient population from a multi-institutional database. Second, a Large Language Model (LLM) is prompted with the retrieved cohort and its associated performance metrics to recommend the optimal prediction algorithm from a pool of eight representative models, including classical linear risk models (e.g., Mayo, Brock), temporally-aware models (e.g., TDVIT, DLSTM), and multi-modal computer vision-based approaches (e.g., Liao, Sybil, DLS, DLI). This two-stage agent pipeline -- retrieval via FAISS and reasoning via LLM -- enables dynamic, cohort-aware risk prediction personalized to each patient's profile. Building on this architecture, the agent supports flexible and cohort-driven model selection across diverse clinical populations, offering a practical path toward individualized risk assessment in real-world lung cancer screening.