 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFlow Matching for Collaborative Filtering
Feb 11, 2025Generative models have shown great promise in collaborative filtering by capturing the underlying distribution of user interests and preferences. However, existing approaches struggle with inaccurate posterior approximations and misalignment with the discrete nature of recommendation data, limiting their expressiveness and real-world performance. To address these limitations, we propose FlowCF, a novel flow-based recommendation system leveraging flow matching for collaborative filtering. We tailor flow matching to the unique challenges in recommendation through two key innovations: (1) a behavior-guided prior that aligns with user behavior patterns to handle the sparse and heterogeneous user-item interactions, and (2) a discrete flow framework to preserve the binary nature of implicit feedback while maintaining the benefits of flow matching, such as stable training and efficient inference. Extensive experiments demonstrate that FlowCF achieves state-of-the-art recommendation accuracy across various datasets with the fastest inference speed, making it a compelling approach for real-world recommender systems.
CaLMFlow: Volterra Flow Matching using Causal Language Models
Oct 03, 2024We introduce CaLMFlow (Causal Language Models for Flow Matching), a novel framework that casts flow matching as a Volterra integral equation (VIE), leveraging the power of large language models (LLMs) for continuous data generation. CaLMFlow enables the direct application of LLMs to learn complex flows by formulating flow matching as a sequence modeling task, bridging discrete language modeling and continuous generative modeling. Our method implements tokenization across space and time, thereby solving a VIE over these domains. This approach enables efficient handling of high-dimensional data and outperforms ODE solver-dependent methods like conditional flow matching (CFM). We demonstrate CaLMFlow's effectiveness on synthetic and real-world data, including single-cell perturbation response prediction, showcasing its ability to incorporate textual context and generalize to unseen conditions. Our results highlight LLM-driven flow matching as a promising paradigm in generative modeling, offering improved scalability, flexibility, and context-awareness.
A Survey on Diffusion Models for Recommender Systems
Sep 08, 2024



While traditional recommendation techniques have made significant strides in the past decades, they still suffer from limited generalization performance caused by factors like inadequate collaborative signals, weak latent representations, and noisy data. In response, diffusion models (DMs) have emerged as promising solutions for recommender systems due to their robust generative capabilities, solid theoretical foundations, and improved training stability. To this end, in this paper, we present the first comprehensive survey on diffusion models for recommendation, and draw a bird's-eye view from the perspective of the whole pipeline in real-world recommender systems. We systematically categorize existing research works into three primary domains: (1) diffusion for data engineering & encoding, focusing on data augmentation and representation enhancement; (2) diffusion as recommender models, employing diffusion models to directly estimate user preferences and rank items; and (3) diffusion for content presentation, utilizing diffusion models to generate personalized content such as fashion and advertisement creatives. Our taxonomy highlights the unique strengths of diffusion models in capturing complex data distributions and generating high-quality, diverse samples that closely align with user preferences. We also summarize the core characteristics of the adapting diffusion models for recommendation, and further identify key areas for future exploration, which helps establish a roadmap for researchers and practitioners seeking to advance recommender systems through the innovative application of diffusion models. To further facilitate the research community of recommender systems based on diffusion models, we actively maintain a GitHub repository for papers and other related resources in this rising direction https://github.com/CHIANGEL/Awesome-Diffusion-for-RecSys.
E3Bind: An End-to-End Equivariant Network for Protein-Ligand Docking
Oct 12, 2022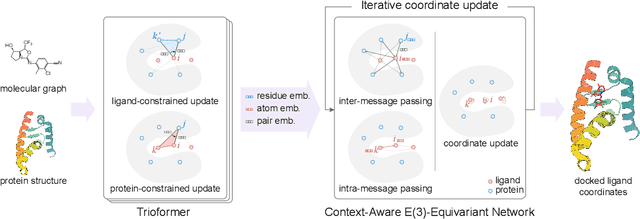
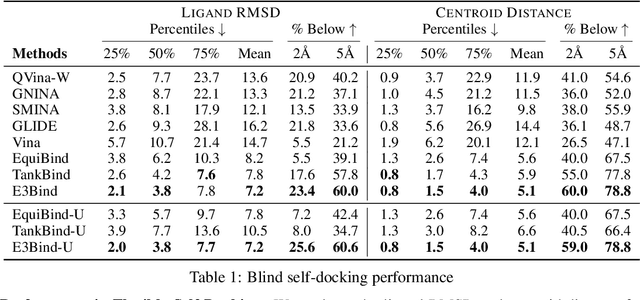
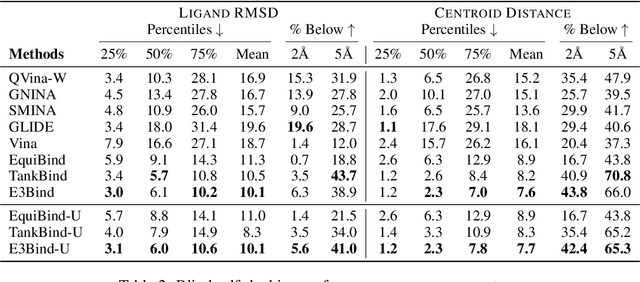
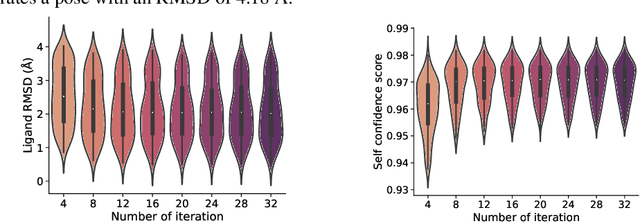
In silico prediction of the ligand binding pose to a given protein target is a crucial but challenging task in drug discovery. This work focuses on blind flexible selfdocking, where we aim to predict the positions, orientations and conformations of docked molecules. Traditional physics-based methods usually suffer from inaccurate scoring functions and high inference cost. Recently, data-driven methods based on deep learning techniques are attracting growing interest thanks to their efficiency during inference and promising performance. These methods usually either adopt a two-stage approach by first predicting the distances between proteins and ligands and then generating the final coordinates based on the predicted distances, or directly predicting the global roto-translation of ligands. In this paper, we take a different route. Inspired by the resounding success of AlphaFold2 for protein structure prediction, we propose E3Bind, an end-to-end equivariant network that iteratively updates the ligand pose. E3Bind models the protein-ligand interaction through careful consideration of the geometric constraints in docking and the local context of the binding site. Experiments on standard benchmark datasets demonstrate the superior performance of our end-to-end trainable model compared to traditional and recently-proposed deep learning methods.
PEER: A Comprehensive and Multi-Task Benchmark for Protein Sequence Understanding
Jun 05, 2022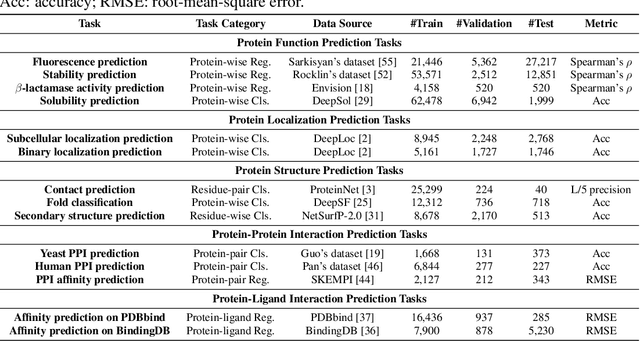
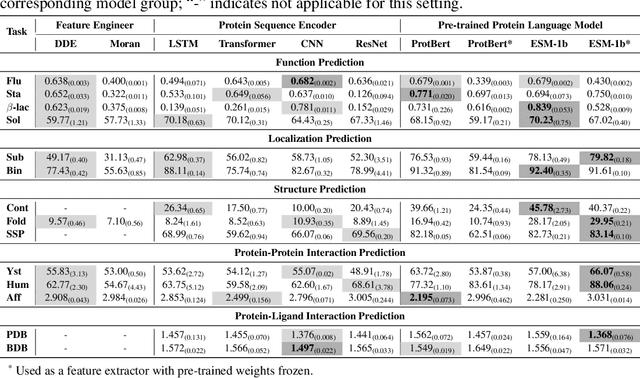

We are now witnessing significant progress of deep learning methods in a variety of tasks (or datasets) of proteins. However, there is a lack of a standard benchmark to evaluate the performance of different methods, which hinders the progress of deep learning in this field. In this paper, we propose such a benchmark called PEER, a comprehensive and multi-task benchmark for Protein sEquence undERstanding. PEER provides a set of diverse protein understanding tasks including protein function prediction, protein localization prediction, protein structure prediction, protein-protein interaction prediction, and protein-ligand interaction prediction. We evaluate different types of sequence-based methods for each task including traditional feature engineering approaches, different sequence encoding methods as well as large-scale pre-trained protein language models. In addition, we also investigate the performance of these methods under the multi-task learning setting. Experimental results show that large-scale pre-trained protein language models achieve the best performance for most individual tasks, and jointly training multiple tasks further boosts the performance. The datasets and source codes of this benchmark will be open-sourced soon.
TorchDrug: A Powerful and Flexible Machine Learning Platform for Drug Discovery
Feb 16, 2022

Machine learning has huge potential to revolutionize the field of drug discovery and is attracting increasing attention in recent years. However, lacking domain knowledge (e.g., which tasks to work on), standard benchmarks and data preprocessing pipelines are the main obstacles for machine learning researchers to work in this domain. To facilitate the progress of machine learning for drug discovery, we develop TorchDrug, a powerful and flexible machine learning platform for drug discovery built on top of PyTorch. TorchDrug benchmarks a variety of important tasks in drug discovery, including molecular property prediction, pretrained molecular representations, de novo molecular design and optimization, retrosynthsis prediction, and biomedical knowledge graph reasoning. State-of-the-art techniques based on geometric deep learning (or graph machine learning), deep generative models, reinforcement learning and knowledge graph reasoning are implemented for these tasks. TorchDrug features a hierarchical interface that facilitates customization from both novices and experts in this domain. Tutorials, benchmark results and documentation are available at https://torchdrug.ai. Code is released under Apache License 2.0.