 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeOGB-LSC: A Large-Scale Challenge for Machine Learning on Graphs
Mar 17, 2021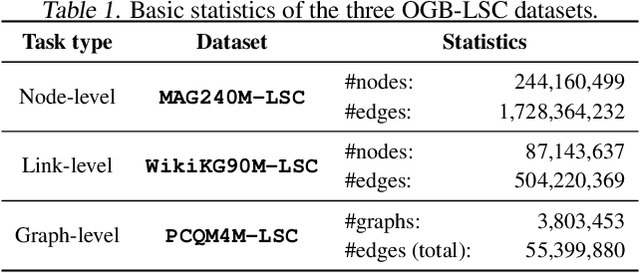
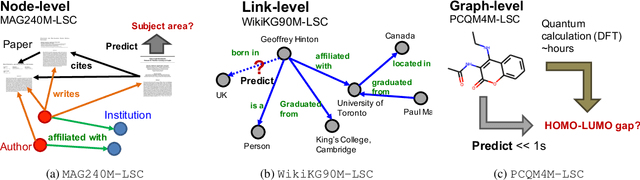
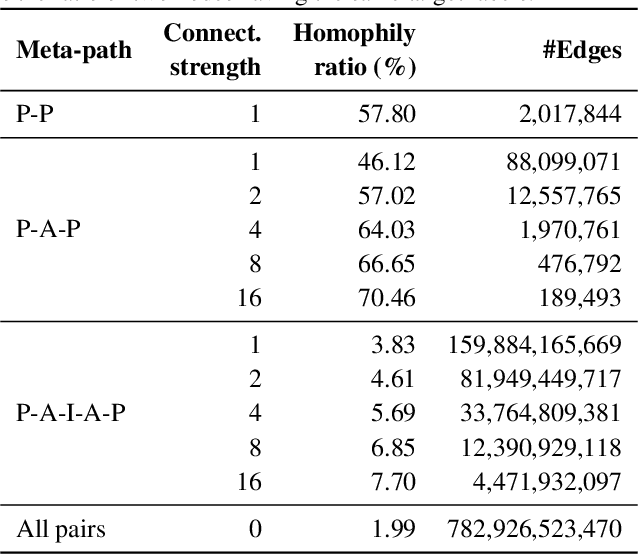
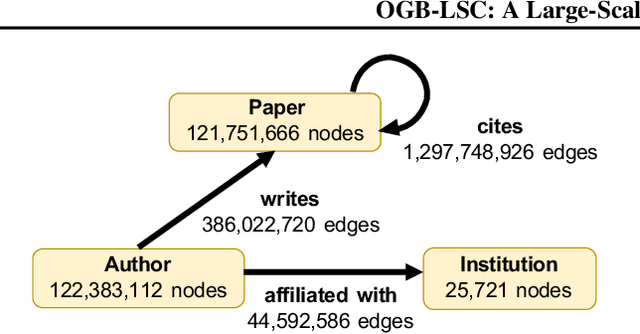
Enabling effective and efficient machine learning (ML) over large-scale graph data (e.g., graphs with billions of edges) can have a huge impact on both industrial and scientific applications. However, community efforts to advance large-scale graph ML have been severely limited by the lack of a suitable public benchmark. For KDD Cup 2021, we present OGB Large-Scale Challenge (OGB-LSC), a collection of three real-world datasets for advancing the state-of-the-art in large-scale graph ML. OGB-LSC provides graph datasets that are orders of magnitude larger than existing ones and covers three core graph learning tasks -- link prediction, graph regression, and node classification. Furthermore, OGB-LSC provides dedicated baseline experiments, scaling up expressive graph ML models to the massive datasets. We show that the expressive models significantly outperform simple scalable baselines, indicating an opportunity for dedicated efforts to further improve graph ML at scale. Our datasets and baseline code are released and maintained as part of our OGB initiative (Hu et al., 2020). We hope OGB-LSC at KDD Cup 2021 can empower the community to discover innovative solutions for large-scale graph ML.
ForceNet: A Graph Neural Network for Large-Scale Quantum Calculations
Mar 02, 2021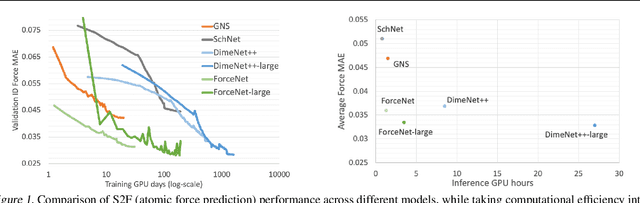
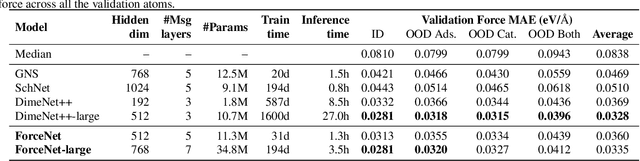
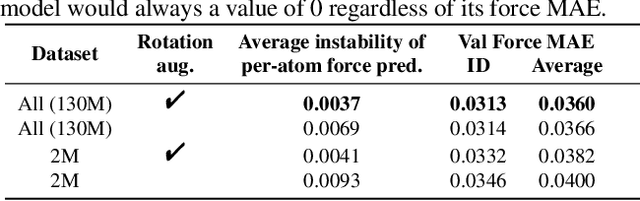
With massive amounts of atomic simulation data available, there is a huge opportunity to develop fast and accurate machine learning models to approximate expensive physics-based calculations. The key quantity to estimate is atomic forces, where the state-of-the-art Graph Neural Networks (GNNs) explicitly enforce basic physical constraints such as rotation-covariance. However, to strictly satisfy the physical constraints, existing models have to make tradeoffs between computational efficiency and model expressiveness. Here we explore an alternative approach. By not imposing explicit physical constraints, we can flexibly design expressive models while maintaining their computational efficiency. Physical constraints are implicitly imposed by training the models using physics-based data augmentation. To evaluate the approach, we carefully design a scalable and expressive GNN model, ForceNet, and apply it to OC20 (Chanussot et al., 2020), an unprecedentedly-large dataset of quantum physics calculations. Our proposed ForceNet is able to predict atomic forces more accurately than state-of-the-art physics-based GNNs while being faster both in training and inference. Overall, our promising and counter-intuitive results open up an exciting avenue for future research.
WILDS: A Benchmark of in-the-Wild Distribution Shifts
Dec 14, 2020
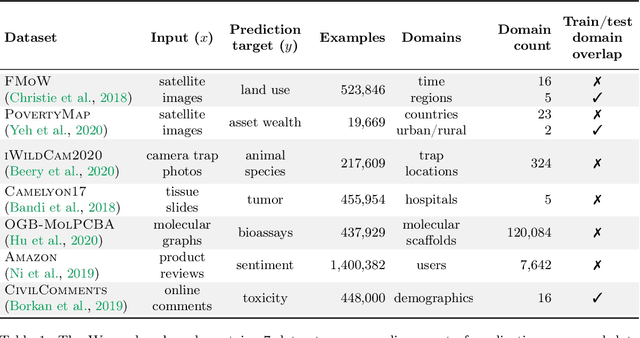

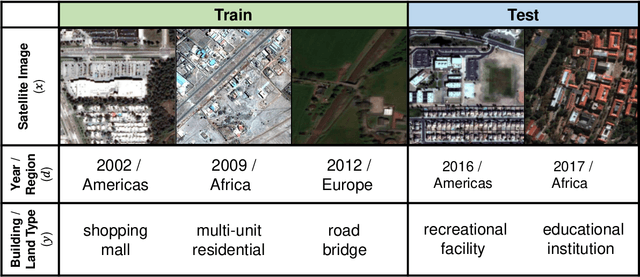
Distribution shifts can cause significant degradation in a broad range of machine learning (ML) systems deployed in the wild. However, many widely-used datasets in the ML community today were not designed for evaluating distribution shifts. These datasets typically have training and test sets drawn from the same distribution, and prior work on retrofitting them with distribution shifts has generally relied on artificial shifts that need not represent the kinds of shifts encountered in the wild. In this paper, we present WILDS, a benchmark of in-the-wild distribution shifts spanning diverse data modalities and applications, from tumor identification to wildlife monitoring to poverty mapping. WILDS builds on top of recent data collection efforts by domain experts in these applications and provides a unified collection of datasets with evaluation metrics and train/test splits that are representative of real-world distribution shifts. These datasets reflect distribution shifts arising from training and testing on different hospitals, cameras, countries, time periods, demographics, molecular scaffolds, etc., all of which cause substantial performance drops in our baseline models. Finally, we survey other applications that would be promising additions to the benchmark but for which we did not manage to find appropriate datasets; we discuss their associated challenges and detail datasets and shifts where we did not see an appreciable performance drop. By unifying datasets from a variety of application areas and making them accessible to the ML community, we hope to encourage the development of general-purpose methods that are anchored to real-world distribution shifts and that work well across different applications and problem settings. Data loaders, default models, and leaderboards are available at https://wilds.stanford.edu.
The Open Catalyst 2020 Dataset and Community Challenges
Oct 20, 2020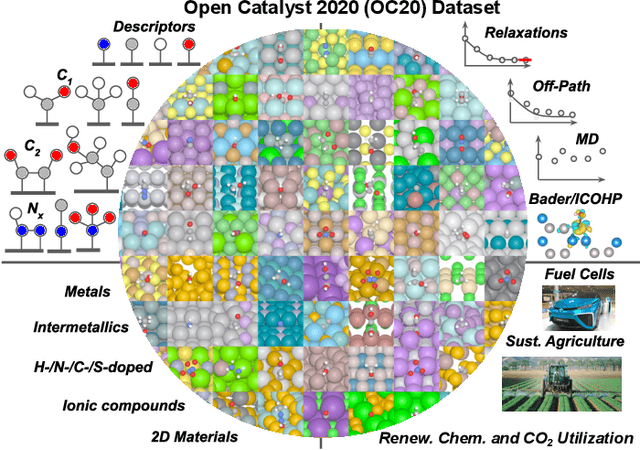

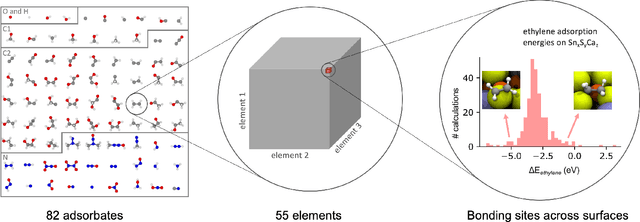
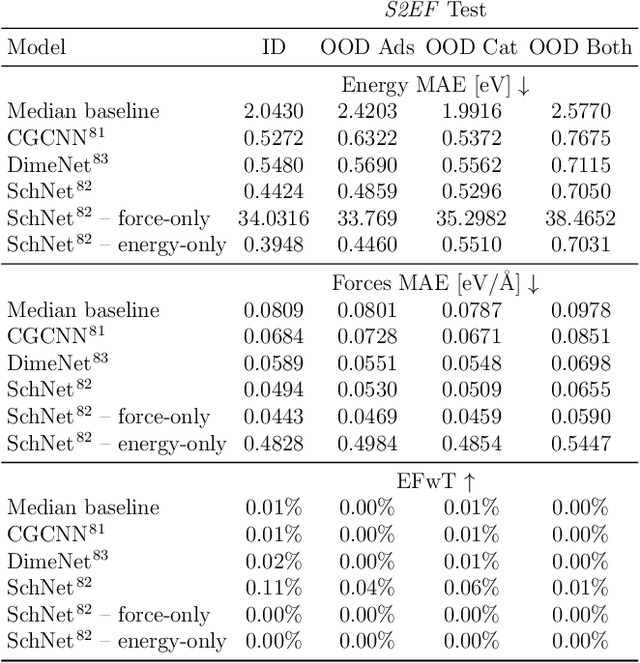
Catalyst discovery and optimization is key to solving many societal and energy challenges including solar fuels synthesis, long-term energy storage, and renewable fertilizer production. Despite considerable effort by the catalysis community to apply machine learning models to the computational catalyst discovery process, it remains an open challenge to build models that can generalize across both elemental compositions of surfaces and adsorbate identity/configurations, perhaps because datasets have been smaller in catalysis than related fields. To address this we developed the OC20 dataset, consisting of 1,281,121 Density Functional Theory (DFT) relaxations (264,900,500 single point evaluations) across a wide swath of materials, surfaces, and adsorbates (nitrogen, carbon, and oxygen chemistries). We supplemented this dataset with randomly perturbed structures, short timescale molecular dynamics, and electronic structure analyses. The dataset comprises three central tasks indicative of day-to-day catalyst modeling and comes with pre-defined train/validation/test splits to facilitate direct comparisons with future model development efforts. We applied three state-of-the-art graph neural network models (SchNet, Dimenet, CGCNN) to each of these tasks as baseline demonstrations for the community to build on. In almost every task, no upper limit on model size was identified, suggesting that even larger models are likely to improve on initial results. The dataset and baseline models are both provided as open resources, as well as a public leader board to encourage community contributions to solve these important tasks.
An Introduction to Electrocatalyst Design using Machine Learning for Renewable Energy Storage
Oct 14, 2020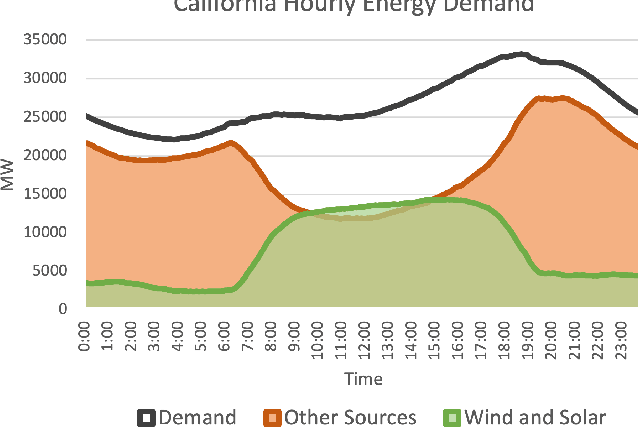
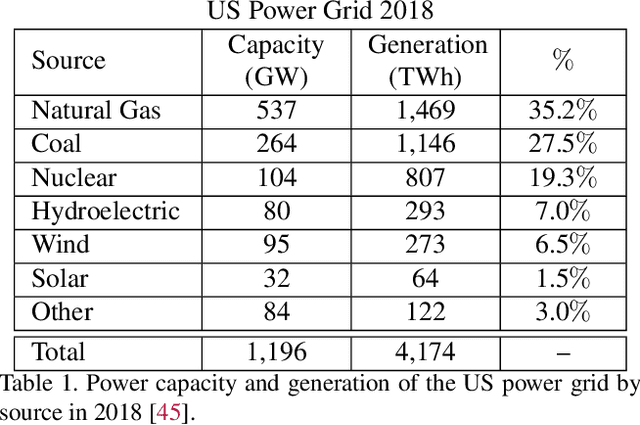
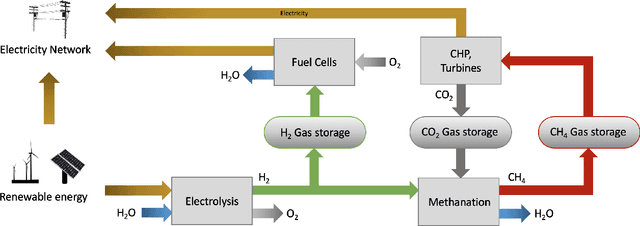
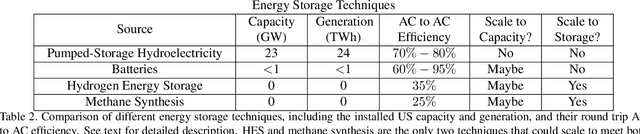
Scalable and cost-effective solutions to renewable energy storage are essential to addressing the world's rising energy needs while reducing climate change. As we increase our reliance on renewable energy sources such as wind and solar, which produce intermittent power, storage is needed to transfer power from times of peak generation to peak demand. This may require the storage of power for hours, days, or months. One solution that offers the potential of scaling to nation-sized grids is the conversion of renewable energy to other fuels, such as hydrogen or methane. To be widely adopted, this process requires cost-effective solutions to running electrochemical reactions. An open challenge is finding low-cost electrocatalysts to drive these reactions at high rates. Through the use of quantum mechanical simulations (density functional theory), new catalyst structures can be tested and evaluated. Unfortunately, the high computational cost of these simulations limits the number of structures that may be tested. The use of machine learning may provide a method to efficiently approximate these calculations, leading to new approaches in finding effective electrocatalysts. In this paper, we provide an introduction to the challenges in finding suitable electrocatalysts, how machine learning may be applied to the problem, and the use of the Open Catalyst Project OC20 dataset for model training.
Open Graph Benchmark: Datasets for Machine Learning on Graphs
May 02, 2020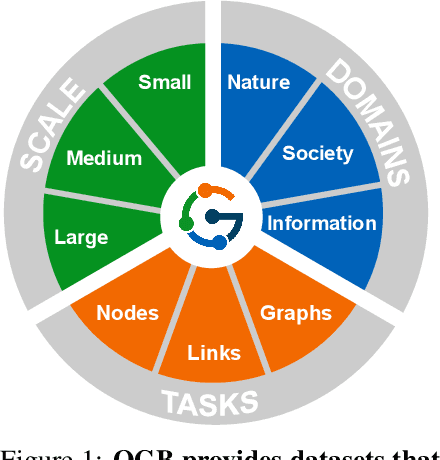
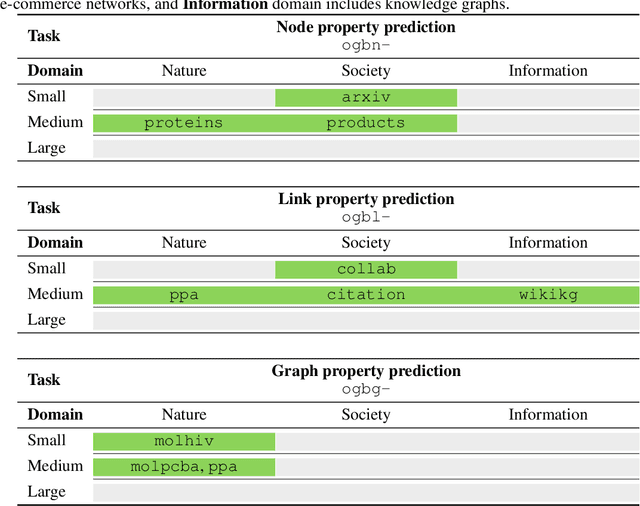
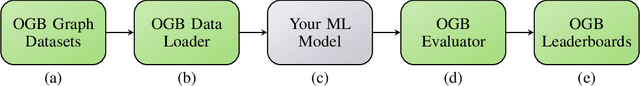
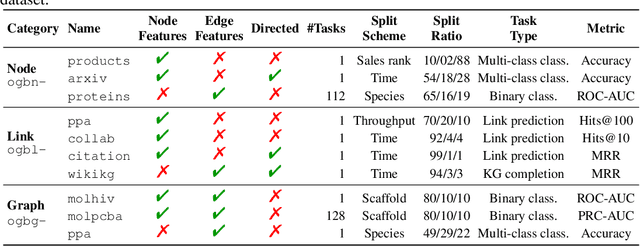
We present the Open Graph Benchmark (OGB), a diverse set of challenging and realistic benchmark datasets to facilitate scalable, robust, and reproducible graph machine learning (ML) research. OGB datasets are large-scale, encompass multiple important graph ML tasks and cover a diverse range of domains, ranging from social and information networks to biological networks, molecular graphs, and knowledge graphs. For each dataset, we provide a unified evaluation protocol using application-specific data splits and evaluation metrics. Our empirical investigation reveals the challenges of existing graph methods in handling large-scale graphs and predicting out-of-distribution data. OGB presents an automated end-to-end graph ML pipeline that simplifies and standardizes the process of graph data loading, experimental setup, and model evaluation. OGB will be regularly updated and welcomes inputs from the community. OGB datasets as well as data loaders and evaluation scripts are available at https://ogb.stanford.edu .
Query2box: Reasoning over Knowledge Graphs in Vector Space using Box Embeddings
Feb 29, 2020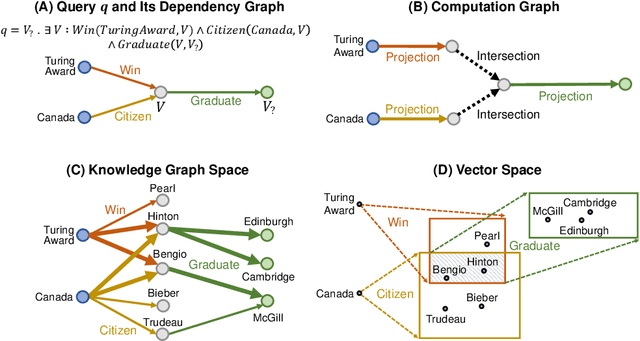
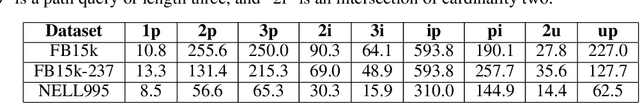
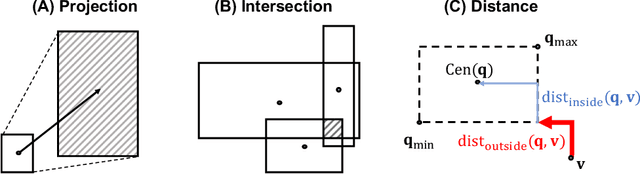
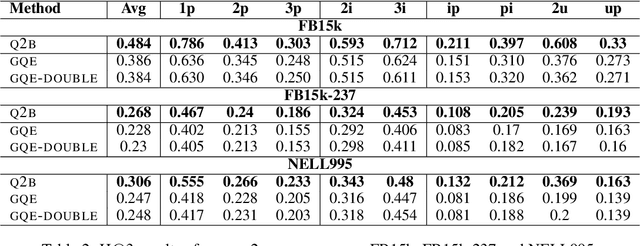
Answering complex logical queries on large-scale incomplete knowledge graphs (KGs) is a fundamental yet challenging task. Recently, a promising approach to this problem has been to embed KG entities as well as the query into a vector space such that entities that answer the query are embedded close to the query. However, prior work models queries as single points in the vector space, which is problematic because a complex query represents a potentially large set of its answer entities, but it is unclear how such a set can be represented as a single point. Furthermore, prior work can only handle queries that use conjunctions ($\wedge$) and existential quantifiers ($\exists$). Handling queries with logical disjunctions ($\vee$) remains an open problem. Here we propose query2box, an embedding-based framework for reasoning over arbitrary queries with $\wedge$, $\vee$, and $\exists$ operators in massive and incomplete KGs. Our main insight is that queries can be embedded as boxes (i.e., hyper-rectangles), where a set of points inside the box corresponds to a set of answer entities of the query. We show that conjunctions can be naturally represented as intersections of boxes and also prove a negative result that handling disjunctions would require embedding with dimension proportional to the number of KG entities. However, we show that by transforming queries into a Disjunctive Normal Form, query2box is capable of handling arbitrary logical queries with $\wedge$, $\vee$, $\exists$ in a scalable manner. We demonstrate the effectiveness of query2box on three large KGs and show that query2box achieves up to 25% relative improvement over the state of the art.
Pre-training Graph Neural Networks
May 29, 2019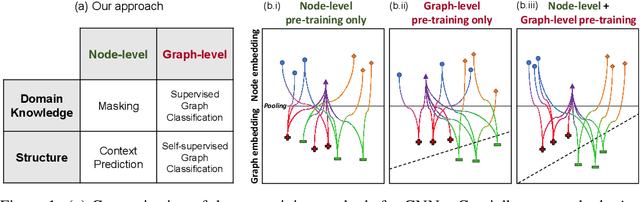
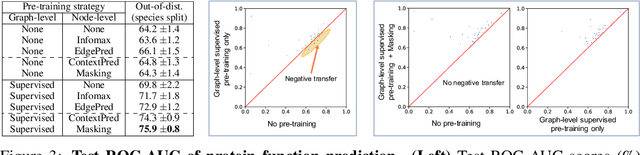
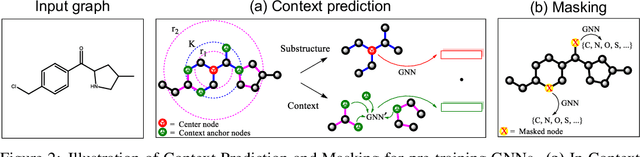
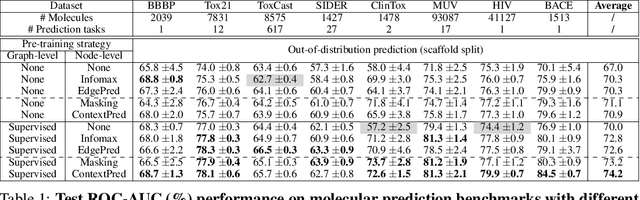
Many applications of machine learning in science and medicine, including molecular property and protein function prediction, can be cast as problems of predicting some properties of graphs, where having good graph representations is critical. However, two key challenges in these domains are (1) extreme scarcity of labeled data due to expensive lab experiments, and (2) needing to extrapolate to test graphs that are structurally different from those seen during training. In this paper, we explore pre-training to address both of these challenges. In particular, working with Graph Neural Networks (GNNs) for representation learning of graphs, we wish to obtain node representations that (1) capture similarity of nodes' network neighborhood structure, (2) can be composed to give accurate graph-level representations, and (3) capture domain-knowledge. To achieve these goals, we propose a series of methods to pre-train GNNs at both the node-level and the graph-level, using both unlabeled data and labeled data from related auxiliary supervised tasks. We perform extensive evaluation on two applications, molecular property and protein function prediction. We observe that performing only graph-level supervised pre-training often leads to marginal performance gain or even can worsen the performance compared to non-pre-trained models. On the other hand, effectively combining both node- and graph-level pre-training techniques significantly improves generalization to out-of-distribution graphs, consistently outperforming non-pre-trained GNNs across 8 datasets in molecular property prediction (resp. 40 tasks in protein function prediction), with the average ROC-AUC improvement of 7.2% (resp. 11.7%).
Co-teaching: Robust Training of Deep Neural Networks with Extremely Noisy Labels
Oct 30, 2018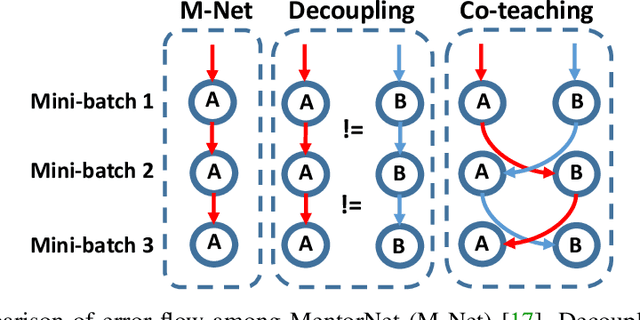
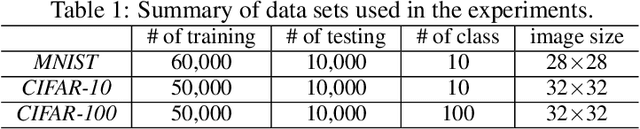
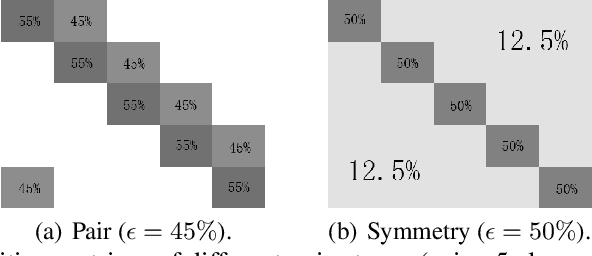
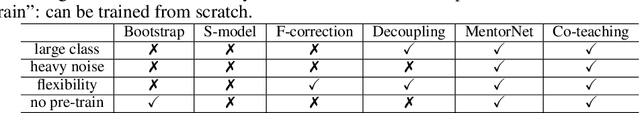
Deep learning with noisy labels is practically challenging, as the capacity of deep models is so high that they can totally memorize these noisy labels sooner or later during training. Nonetheless, recent studies on the memorization effects of deep neural networks show that they would first memorize training data of clean labels and then those of noisy labels. Therefore in this paper, we propose a new deep learning paradigm called Co-teaching for combating with noisy labels. Namely, we train two deep neural networks simultaneously, and let them teach each other given every mini-batch: firstly, each network feeds forward all data and selects some data of possibly clean labels; secondly, two networks communicate with each other what data in this mini-batch should be used for training; finally, each network back propagates the data selected by its peer network and updates itself. Empirical results on noisy versions of MNIST, CIFAR-10 and CIFAR-100 demonstrate that Co-teaching is much superior to the state-of-the-art methods in the robustness of trained deep models.
How Powerful are Graph Neural Networks?
Oct 01, 2018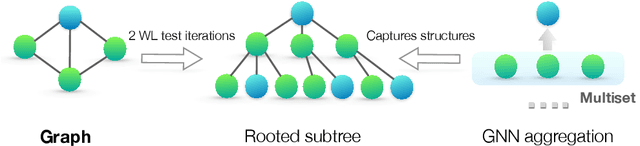
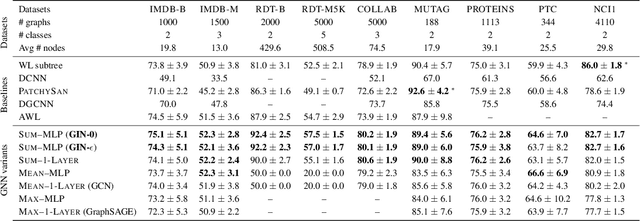

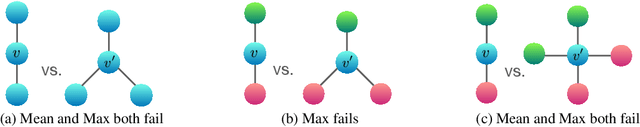
Graph Neural Networks (GNNs) for representation learning of graphs broadly follow a neighborhood aggregation framework, where the representation vector of a node is computed by recursively aggregating and transforming feature vectors of its neighboring nodes. Many GNN variants have been proposed and have achieved state-of-the-art results on both node and graph classification tasks. However, despite GNNs revolutionizing graph representation learning, there is limited understanding of their representational properties and limitations. Here, we present a theoretical framework for analyzing the expressive power of GNNs in capturing different graph structures. Our results characterize the discriminative power of popular GNN variants, such as Graph Convolutional Networks and GraphSAGE, and show that they cannot learn to distinguish certain simple graph structures. We then develop a simple architecture that is provably the most expressive among the class of GNNs and is as powerful as the Weisfeiler-Lehman graph isomorphism test. We empirically validate our theoretical findings on a number of graph classification benchmarks, and demonstrate that our model achieves state-of-the-art performance.