 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeThe Open Catalyst 2020 Dataset and Community Challenges
Oct 20, 2020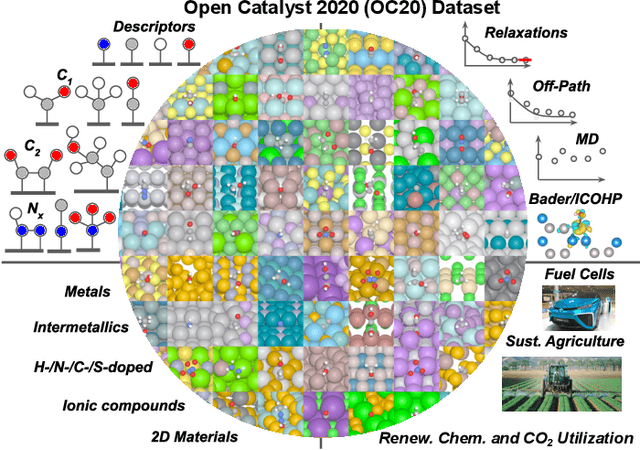

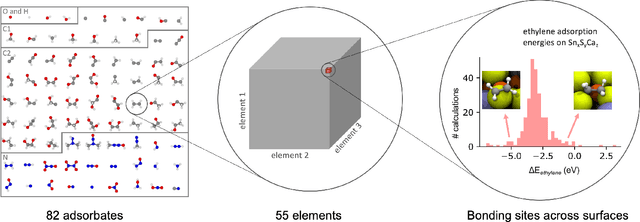
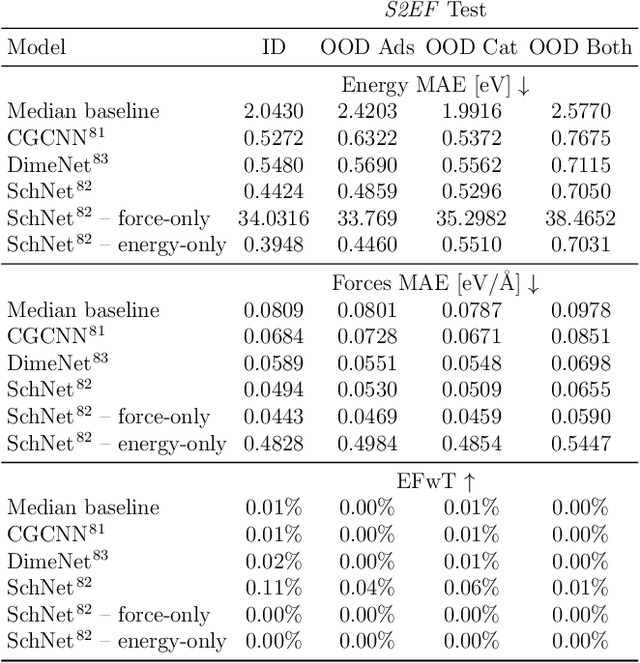
Catalyst discovery and optimization is key to solving many societal and energy challenges including solar fuels synthesis, long-term energy storage, and renewable fertilizer production. Despite considerable effort by the catalysis community to apply machine learning models to the computational catalyst discovery process, it remains an open challenge to build models that can generalize across both elemental compositions of surfaces and adsorbate identity/configurations, perhaps because datasets have been smaller in catalysis than related fields. To address this we developed the OC20 dataset, consisting of 1,281,121 Density Functional Theory (DFT) relaxations (264,900,500 single point evaluations) across a wide swath of materials, surfaces, and adsorbates (nitrogen, carbon, and oxygen chemistries). We supplemented this dataset with randomly perturbed structures, short timescale molecular dynamics, and electronic structure analyses. The dataset comprises three central tasks indicative of day-to-day catalyst modeling and comes with pre-defined train/validation/test splits to facilitate direct comparisons with future model development efforts. We applied three state-of-the-art graph neural network models (SchNet, Dimenet, CGCNN) to each of these tasks as baseline demonstrations for the community to build on. In almost every task, no upper limit on model size was identified, suggesting that even larger models are likely to improve on initial results. The dataset and baseline models are both provided as open resources, as well as a public leader board to encourage community contributions to solve these important tasks.
An Introduction to Electrocatalyst Design using Machine Learning for Renewable Energy Storage
Oct 14, 2020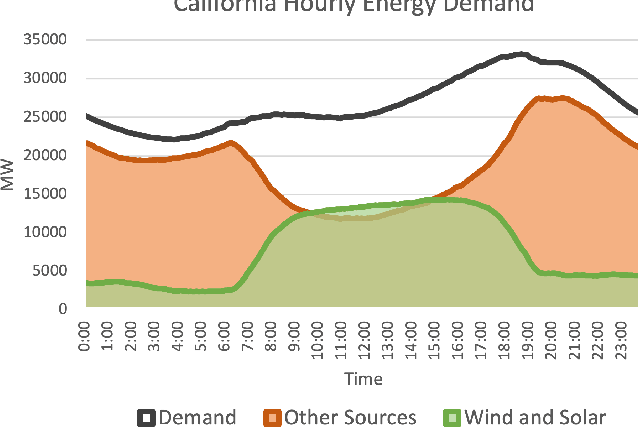
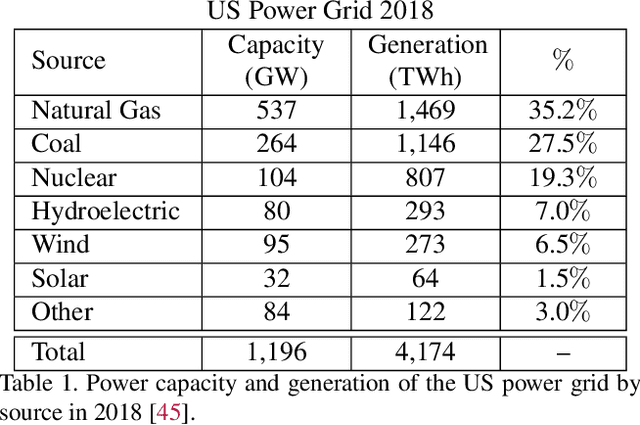
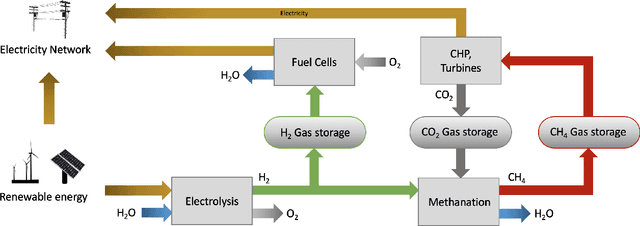
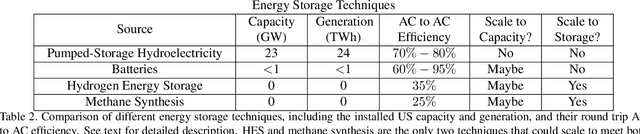
Scalable and cost-effective solutions to renewable energy storage are essential to addressing the world's rising energy needs while reducing climate change. As we increase our reliance on renewable energy sources such as wind and solar, which produce intermittent power, storage is needed to transfer power from times of peak generation to peak demand. This may require the storage of power for hours, days, or months. One solution that offers the potential of scaling to nation-sized grids is the conversion of renewable energy to other fuels, such as hydrogen or methane. To be widely adopted, this process requires cost-effective solutions to running electrochemical reactions. An open challenge is finding low-cost electrocatalysts to drive these reactions at high rates. Through the use of quantum mechanical simulations (density functional theory), new catalyst structures can be tested and evaluated. Unfortunately, the high computational cost of these simulations limits the number of structures that may be tested. The use of machine learning may provide a method to efficiently approximate these calculations, leading to new approaches in finding effective electrocatalysts. In this paper, we provide an introduction to the challenges in finding suitable electrocatalysts, how machine learning may be applied to the problem, and the use of the Open Catalyst Project OC20 dataset for model training.