 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMarco-Bench-MIF: On Multilingual Instruction-Following Capability of Large Language Models
Jul 16, 2025Instruction-following capability has become a major ability to be evaluated for Large Language Models (LLMs). However, existing datasets, such as IFEval, are either predominantly monolingual and centered on English or simply machine translated to other languages, limiting their applicability in multilingual contexts. In this paper, we present an carefully-curated extension of IFEval to a localized multilingual version named Marco-Bench-MIF, covering 30 languages with varying levels of localization. Our benchmark addresses linguistic constraints (e.g., modifying capitalization requirements for Chinese) and cultural references (e.g., substituting region-specific company names in prompts) via a hybrid pipeline combining translation with verification. Through comprehensive evaluation of 20+ LLMs on our Marco-Bench-MIF, we found that: (1) 25-35% accuracy gap between high/low-resource languages, (2) model scales largely impact performance by 45-60% yet persists script-specific challenges, and (3) machine-translated data underestimates accuracy by7-22% versus localized data. Our analysis identifies challenges in multilingual instruction following, including keyword consistency preservation and compositional constraint adherence across languages. Our Marco-Bench-MIF is available at https://github.com/AIDC-AI/Marco-Bench-MIF.
The Bitter Lesson Learned from 2,000+ Multilingual Benchmarks
Apr 22, 2025



As large language models (LLMs) continue to advance in linguistic capabilities, robust multilingual evaluation has become essential for promoting equitable technological progress. This position paper examines over 2,000 multilingual (non-English) benchmarks from 148 countries, published between 2021 and 2024, to evaluate past, present, and future practices in multilingual benchmarking. Our findings reveal that, despite significant investments amounting to tens of millions of dollars, English remains significantly overrepresented in these benchmarks. Additionally, most benchmarks rely on original language content rather than translations, with the majority sourced from high-resource countries such as China, India, Germany, the UK, and the USA. Furthermore, a comparison of benchmark performance with human judgments highlights notable disparities. STEM-related tasks exhibit strong correlations with human evaluations (0.70 to 0.85), while traditional NLP tasks like question answering (e.g., XQuAD) show much weaker correlations (0.11 to 0.30). Moreover, translating English benchmarks into other languages proves insufficient, as localized benchmarks demonstrate significantly higher alignment with local human judgments (0.68) than their translated counterparts (0.47). This underscores the importance of creating culturally and linguistically tailored benchmarks rather than relying solely on translations. Through this comprehensive analysis, we highlight six key limitations in current multilingual evaluation practices, propose the guiding principles accordingly for effective multilingual benchmarking, and outline five critical research directions to drive progress in the field. Finally, we call for a global collaborative effort to develop human-aligned benchmarks that prioritize real-world applications.
New Trends for Modern Machine Translation with Large Reasoning Models
Mar 13, 2025



Recent advances in Large Reasoning Models (LRMs), particularly those leveraging Chain-of-Thought reasoning (CoT), have opened brand new possibility for Machine Translation (MT). This position paper argues that LRMs substantially transformed traditional neural MT as well as LLMs-based MT paradigms by reframing translation as a dynamic reasoning task that requires contextual, cultural, and linguistic understanding and reasoning. We identify three foundational shifts: 1) contextual coherence, where LRMs resolve ambiguities and preserve discourse structure through explicit reasoning over cross-sentence and complex context or even lack of context; 2) cultural intentionality, enabling models to adapt outputs by inferring speaker intent, audience expectations, and socio-linguistic norms; 3) self-reflection, LRMs can perform self-reflection during the inference time to correct the potential errors in translation especially extremely noisy cases, showing better robustness compared to simply mapping X->Y translation. We explore various scenarios in translation including stylized translation, document-level translation and multimodal translation by showcasing empirical examples that demonstrate the superiority of LRMs in translation. We also identify several interesting phenomenons for LRMs for MT including auto-pivot translation as well as the critical challenges such as over-localisation in translation and inference efficiency. In conclusion, we think that LRMs redefine translation systems not merely as text converters but as multilingual cognitive agents capable of reasoning about meaning beyond the text. This paradigm shift reminds us to think of problems in translation beyond traditional translation scenarios in a much broader context with LRMs - what we can achieve on top of it.
An End-to-End Model for Photo-Sharing Multi-modal Dialogue Generation
Aug 16, 2024



Photo-Sharing Multi-modal dialogue generation requires a dialogue agent not only to generate text responses but also to share photos at the proper moment. Using image text caption as the bridge, a pipeline model integrates an image caption model, a text generation model, and an image generation model to handle this complex multi-modal task. However, representing the images with text captions may loss important visual details and information and cause error propagation in the complex dialogue system. Besides, the pipeline model isolates the three models separately because discrete image text captions hinder end-to-end gradient propagation. We propose the first end-to-end model for photo-sharing multi-modal dialogue generation, which integrates an image perceptron and an image generator with a large language model. The large language model employs the Q-Former to perceive visual images in the input end. For image generation in the output end, we propose a dynamic vocabulary transformation matrix and use straight-through and gumbel-softmax techniques to align the large language model and stable diffusion model and achieve end-to-end gradient propagation. We perform experiments on PhotoChat and DialogCC datasets to evaluate our end-to-end model. Compared with pipeline models, the end-to-end model gains state-of-the-art performances on various metrics of text and image generation. More analysis experiments also verify the effectiveness of the end-to-end model for photo-sharing multi-modal dialogue generation.
SHREC 2021: Classification in cryo-electron tomograms
Mar 18, 2022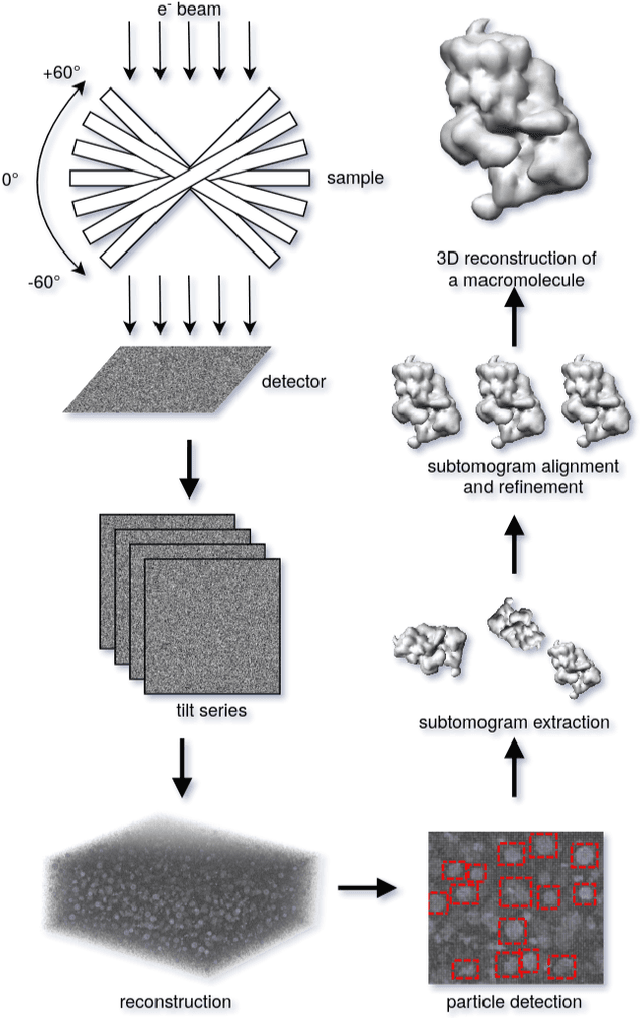
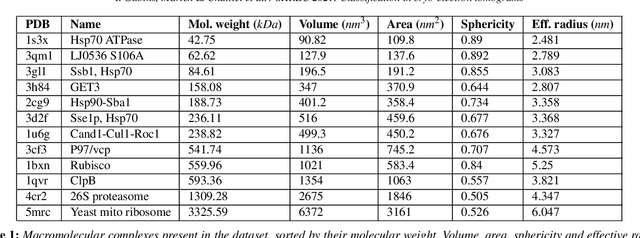
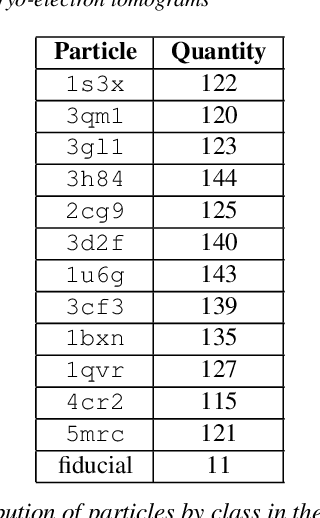
Cryo-electron tomography (cryo-ET) is an imaging technique that allows three-dimensional visualization of macro-molecular assemblies under near-native conditions. Cryo-ET comes with a number of challenges, mainly low signal-to-noise and inability to obtain images from all angles. Computational methods are key to analyze cryo-electron tomograms. To promote innovation in computational methods, we generate a novel simulated dataset to benchmark different methods of localization and classification of biological macromolecules in tomograms. Our publicly available dataset contains ten tomographic reconstructions of simulated cell-like volumes. Each volume contains twelve different types of complexes, varying in size, function and structure. In this paper, we have evaluated seven different methods of finding and classifying proteins. Seven research groups present results obtained with learning-based methods and trained on the simulated dataset, as well as a baseline template matching (TM), a traditional method widely used in cryo-ET research. We show that learning-based approaches can achieve notably better localization and classification performance than TM. We also experimentally confirm that there is a negative relationship between particle size and performance for all methods.
Cryo-shift: Reducing domain shift in cryo-electron subtomograms with unsupervised domain adaptation and randomization
Nov 17, 2021

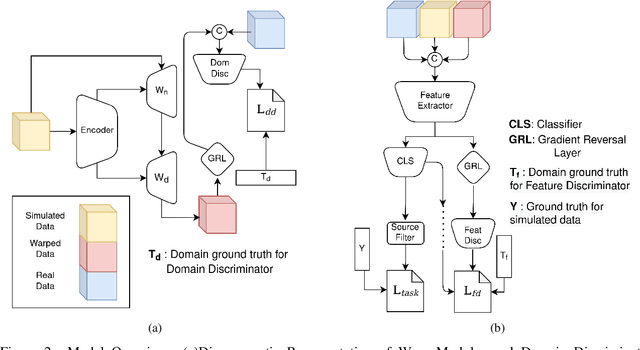

Cryo-Electron Tomography (cryo-ET) is a 3D imaging technology that enables the visualization of subcellular structures in situ at near-atomic resolution. Cellular cryo-ET images help in resolving the structures of macromolecules and determining their spatial relationship in a single cell, which has broad significance in cell and structural biology. Subtomogram classification and recognition constitute a primary step in the systematic recovery of these macromolecular structures. Supervised deep learning methods have been proven to be highly accurate and efficient for subtomogram classification, but suffer from limited applicability due to scarcity of annotated data. While generating simulated data for training supervised models is a potential solution, a sizeable difference in the image intensity distribution in generated data as compared to real experimental data will cause the trained models to perform poorly in predicting classes on real subtomograms. In this work, we present Cryo-Shift, a fully unsupervised domain adaptation and randomization framework for deep learning-based cross-domain subtomogram classification. We use unsupervised multi-adversarial domain adaption to reduce the domain shift between features of simulated and experimental data. We develop a network-driven domain randomization procedure with `warp' modules to alter the simulated data and help the classifier generalize better on experimental data. We do not use any labeled experimental data to train our model, whereas some of the existing alternative approaches require labeled experimental samples for cross-domain classification. Nevertheless, Cryo-Shift outperforms the existing alternative approaches in cross-domain subtomogram classification in extensive evaluation studies demonstrated herein using both simulated and experimental data.
* 14 pages