 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAgents' Last Exam
Jun 03, 2026Recent AI systems have achieved strong results on a wide range of benchmarks, yet these gains have not translated into economically meaningful deployment across many professional domains. We argue that this gap is largely an evaluation problem: widely used benchmarks lack sustained performance measurement on real and economically valuable workflows. This paper introduces Agents' Last Exam (ALE), a benchmark designed to evaluate AI agents on long-horizon, economically valuable, real-world tasks with verifiable outcomes. Developed in collaboration with 250+ industry experts, ALE covers non-physical industries defined with reference to O*NET / SOC 2018 (the U.S. federal occupational taxonomy). It is organized around a task taxonomy with 55 subfields grouped into 13 industry clusters covering 1K+ tasks. Current results show that the hardest tier remains far from saturated: across mainstream harness and backbone configurations, the average full pass rate is 2.6%. ALE is designed as a living benchmark: its task pool grows continuously as new workflows and industries are onboarded. More broadly, ALE is intended not merely as another leaderboard, but as an instrument for closing the gap between benchmark success and GDP-relevant impact.
Enabling Ultra-Fast Cardiovascular Imaging Across Heterogeneous Clinical Environments with a Generalist Foundation Model and Multimodal Database
Dec 25, 2025Multimodal cardiovascular magnetic resonance (CMR) imaging provides comprehensive and non-invasive insights into cardiovascular disease (CVD) diagnosis and underlying mechanisms. Despite decades of advancements, its widespread clinical adoption remains constrained by prolonged scan times and heterogeneity across medical environments. This underscores the urgent need for a generalist reconstruction foundation model for ultra-fast CMR imaging, one capable of adapting across diverse imaging scenarios and serving as the essential substrate for all downstream analyses. To enable this goal, we curate MMCMR-427K, the largest and most comprehensive multimodal CMR k-space database to date, comprising 427,465 multi-coil k-space data paired with structured metadata across 13 international centers, 12 CMR modalities, 15 scanners, and 17 CVD categories in populations across three continents. Building on this unprecedented resource, we introduce CardioMM, a generalist reconstruction foundation model capable of dynamically adapting to heterogeneous fast CMR imaging scenarios. CardioMM unifies semantic contextual understanding with physics-informed data consistency to deliver robust reconstructions across varied scanners, protocols, and patient presentations. Comprehensive evaluations demonstrate that CardioMM achieves state-of-the-art performance in the internal centers and exhibits strong zero-shot generalization to unseen external settings. Even at imaging acceleration up to 24x, CardioMM reliably preserves key cardiac phenotypes, quantitative myocardial biomarkers, and diagnostic image quality, enabling a substantial increase in CMR examination throughput without compromising clinical integrity. Together, our open-access MMCMR-427K database and CardioMM framework establish a scalable pathway toward high-throughput, high-quality, and clinically accessible cardiovascular imaging.
DiffKnock: Diffusion-based Knockoff Statistics for Neural Networks Inference
Oct 01, 2025We introduce DiffKnock, a diffusion-based knockoff framework for high-dimensional feature selection with finite-sample false discovery rate (FDR) control. DiffKnock addresses two key limitations of existing knockoff methods: preserving complex feature dependencies and detecting non-linear associations. Our approach trains diffusion models to generate valid knockoffs and uses neural network--based gradient and filter statistics to construct antisymmetric feature importance measures. Through simulations, we showed that DiffKnock achieved higher power than autoencoder-based knockoffs while maintaining target FDR, indicating its superior performance in scenarios involving complex non-linear architectures. Applied to murine single-cell RNA-seq data of LPS-stimulated macrophages, DiffKnock identifies canonical NF-$\kappa$B target genes (Ccl3, Hmox1) and regulators (Fosb, Pdgfb). These results highlight that, by combining the flexibility of deep generative models with rigorous statistical guarantees, DiffKnock is a powerful and reliable tool for analyzing single-cell RNA-seq data, as well as high-dimensional and structured data in other domains.
A Generalized Genetic Random Field Method for the Genetic Association Analysis of Sequencing Data
Aug 18, 2025With the advance of high-throughput sequencing technologies, it has become feasible to investigate the influence of the entire spectrum of sequencing variations on complex human diseases. Although association studies utilizing the new sequencing technologies hold great promise to unravel novel genetic variants, especially rare genetic variants that contribute to human diseases, the statistical analysis of high-dimensional sequencing data remains a challenge. Advanced analytical methods are in great need to facilitate high-dimensional sequencing data analyses. In this article, we propose a generalized genetic random field (GGRF) method for association analyses of sequencing data. Like other similarity-based methods (e.g., SIMreg and SKAT), the new method has the advantages of avoiding the need to specify thresholds for rare variants and allowing for testing multiple variants acting in different directions and magnitude of effects. The method is built on the generalized estimating equation framework and thus accommodates a variety of disease phenotypes (e.g., quantitative and binary phenotypes). Moreover, it has a nice asymptotic property, and can be applied to small-scale sequencing data without need for small-sample adjustment. Through simulations, we demonstrate that the proposed GGRF attains an improved or comparable power over a commonly used method, SKAT, under various disease scenarios, especially when rare variants play a significant role in disease etiology. We further illustrate GGRF with an application to a real dataset from the Dallas Heart Study. By using GGRF, we were able to detect the association of two candidate genes, ANGPTL3 and ANGPTL4, with serum triglyceride.
A Kernel-Based Neural Network Test for High-dimensional Sequencing Data Analysis
Dec 06, 2023The recent development of artificial intelligence (AI) technology, especially the advance of deep neural network (DNN) technology, has revolutionized many fields. While DNN plays a central role in modern AI technology, it has been rarely used in sequencing data analysis due to challenges brought by high-dimensional sequencing data (e.g., overfitting). Moreover, due to the complexity of neural networks and their unknown limiting distributions, building association tests on neural networks for genetic association analysis remains a great challenge. To address these challenges and fill the important gap of using AI in high-dimensional sequencing data analysis, we introduce a new kernel-based neural network (KNN) test for complex association analysis of sequencing data. The test is built on our previously developed KNN framework, which uses random effects to model the overall effects of high-dimensional genetic data and adopts kernel-based neural network structures to model complex genotype-phenotype relationships. Based on KNN, a Wald-type test is then introduced to evaluate the joint association of high-dimensional genetic data with a disease phenotype of interest, considering non-linear and non-additive effects (e.g., interaction effects). Through simulations, we demonstrated that our proposed method attained higher power compared to the sequence kernel association test (SKAT), especially in the presence of non-linear and interaction effects. Finally, we apply the methods to the whole genome sequencing (WGS) dataset from the Alzheimer's Disease Neuroimaging Initiative (ADNI) study, investigating new genes associated with the hippocampal volume change over time.
An Association Test Based on Kernel-Based Neural Networks for Complex Genetic Association Analysis
Dec 06, 2023The advent of artificial intelligence, especially the progress of deep neural networks, is expected to revolutionize genetic research and offer unprecedented potential to decode the complex relationships between genetic variants and disease phenotypes, which could mark a significant step toward improving our understanding of the disease etiology. While deep neural networks hold great promise for genetic association analysis, limited research has been focused on developing neural-network-based tests to dissect complex genotype-phenotype associations. This complexity arises from the opaque nature of neural networks and the absence of defined limiting distributions. We have previously developed a kernel-based neural network model (KNN) that synergizes the strengths of linear mixed models with conventional neural networks. KNN adopts a computationally efficient minimum norm quadratic unbiased estimator (MINQUE) algorithm and uses KNN structure to capture the complex relationship between large-scale sequencing data and a disease phenotype of interest. In the KNN framework, we introduce a MINQUE-based test to assess the joint association of genetic variants with the phenotype, which considers non-linear and non-additive effects and follows a mixture of chi-square distributions. We also construct two additional tests to evaluate and interpret linear and non-linear/non-additive genetic effects, including interaction effects. Our simulations show that our method consistently controls the type I error rate under various conditions and achieves greater power than a commonly used sequence kernel association test (SKAT), especially when involving non-linear and interaction effects. When applied to real data from the UK Biobank, our approach identified genes associated with hippocampal volume, which can be further replicated and evaluated for their role in the pathogenesis of Alzheimer's disease.
Proof-of-Federated-Learning-Subchain: Free Partner Selection Subchain Based on Federated Learning
Jul 30, 2023



The continuous thriving of the Blockchain society motivates research in novel designs of schemes supporting cryptocurrencies. Previously multiple Proof-of-Deep-Learning(PoDL) consensuses have been proposed to replace hashing with useful work such as deep learning model training tasks. The energy will be more efficiently used while maintaining the ledger. However deep learning models are problem-specific and can be extremely complex. Current PoDL consensuses still require much work to realize in the real world. In this paper, we proposed a novel consensus named Proof-of-Federated-Learning-Subchain(PoFLSC) to fill the gap. We applied a subchain to record the training, challenging, and auditing activities and emphasized the importance of valuable datasets in partner selection. We simulated 20 miners in the subchain to demonstrate the effectiveness of PoFLSC. When we reduce the pool size concerning the reservation priority order, the drop rate difference in the performance in different scenarios further exhibits that the miner with a higher Shapley Value (SV) will gain a better opportunity to be selected when the size of the subchain pool is limited. In the conducted experiments, the PoFLSC consensus supported the subchain manager to be aware of reservation priority and the core partition of contributors to establish and maintain a competitive subchain.
A Sieve Quasi-likelihood Ratio Test for Neural Networks with Applications to Genetic Association Studies
Dec 16, 2022Neural networks (NN) play a central role in modern Artificial intelligence (AI) technology and has been successfully used in areas such as natural language processing and image recognition. While majority of NN applications focus on prediction and classification, there are increasing interests in studying statistical inference of neural networks. The study of NN statistical inference can enhance our understanding of NN statistical proprieties. Moreover, it can facilitate the NN-based hypothesis testing that can be applied to hypothesis-driven clinical and biomedical research. In this paper, we propose a sieve quasi-likelihood ratio test based on NN with one hidden layer for testing complex associations. The test statistic has asymptotic chi-squared distribution, and therefore it is computationally efficient and easy for implementation in real data analysis. The validity of the asymptotic distribution is investigated via simulations. Finally, we demonstrate the use of the proposed test by performing a genetic association analysis of the sequencing data from Alzheimer's Disease Neuroimaging Initiative (ADNI).
A Neural Network Based Method with Transfer Learning for Genetic Data Analysis
Jun 20, 2022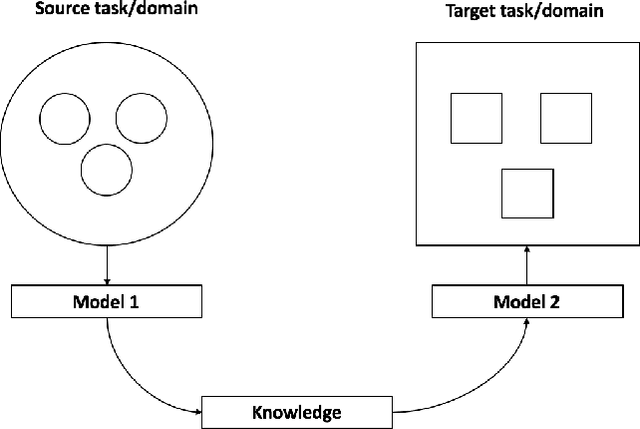
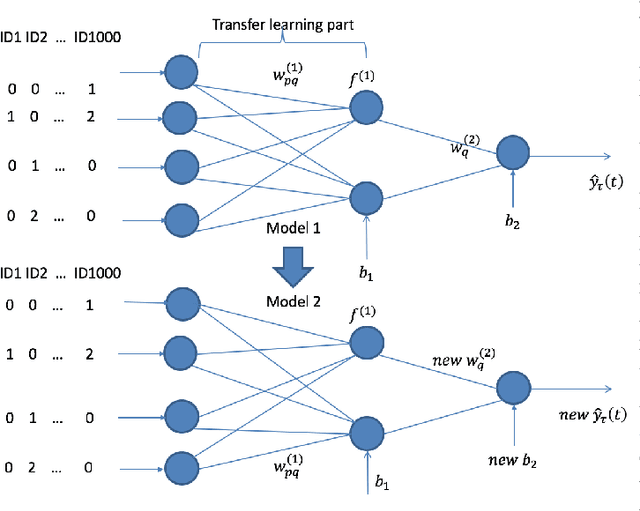
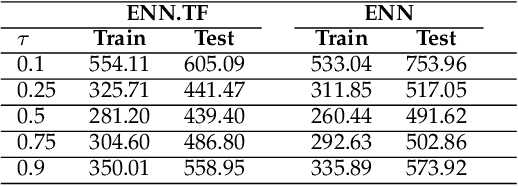
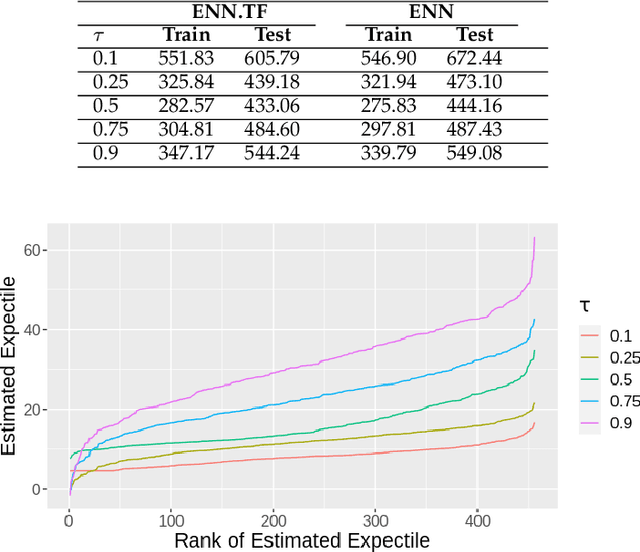
Transfer learning has emerged as a powerful technique in many application problems, such as computer vision and natural language processing. However, this technique is largely ignored in application to genetic data analysis. In this paper, we combine transfer learning technique with a neural network based method(expectile neural networks). With transfer learning, instead of starting the learning process from scratch, we start from one task that have been learned when solving a different task. We leverage previous learnings and avoid starting from scratch to improve the model performance by passing information gained in different but related task. To demonstrate the performance, we run two real data sets. By using transfer learning algorithm, the performance of expectile neural networks is improved compared to expectile neural network without using transfer learning technique.
RT-DNAS: Real-time Constrained Differentiable Neural Architecture Search for 3D Cardiac Cine MRI Segmentation
Jun 13, 2022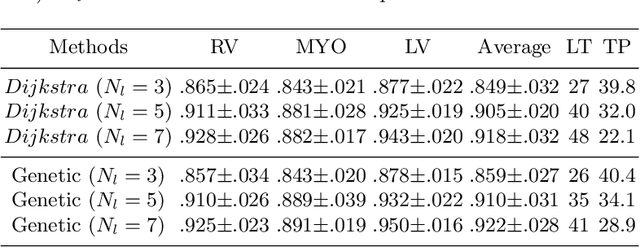

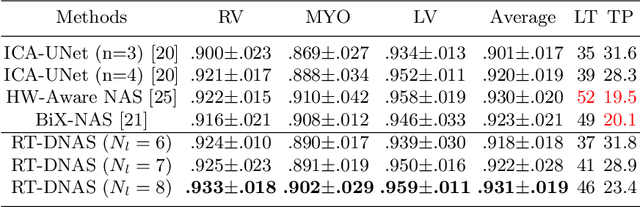
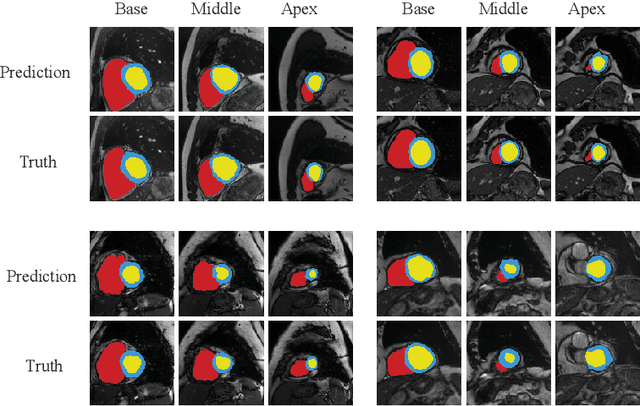
Accurately segmenting temporal frames of cine magnetic resonance imaging (MRI) is a crucial step in various real-time MRI guided cardiac interventions. To achieve fast and accurate visual assistance, there are strict requirements on the maximum latency and minimum throughput of the segmentation framework. State-of-the-art neural networks on this task are mostly hand-crafted to satisfy these constraints while achieving high accuracy. On the other hand, while existing literature have demonstrated the power of neural architecture search (NAS) in automatically identifying the best neural architectures for various medical applications, they are mostly guided by accuracy, sometimes with computation complexity, and the importance of real-time constraints are overlooked. A major challenge is that such constraints are non-differentiable and are thus not compatible with the widely used differentiable NAS frameworks. In this paper, we present a strategy that directly handles real-time constraints in a differentiable NAS framework named RT-DNAS. Experiments on extended 2017 MICCAI ACDC dataset show that compared with state-of-the-art manually and automatically designed architectures, RT-DNAS is able to identify ones with better accuracy while satisfying the real-time constraints.