 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeBeyond Benchmark: LLMs Evaluation with an Anthropomorphic and Value-oriented Roadmap
Aug 26, 2025For Large Language Models (LLMs), a disconnect persists between benchmark performance and real-world utility. Current evaluation frameworks remain fragmented, prioritizing technical metrics while neglecting holistic assessment for deployment. This survey introduces an anthropomorphic evaluation paradigm through the lens of human intelligence, proposing a novel three-dimensional taxonomy: Intelligence Quotient (IQ)-General Intelligence for foundational capacity, Emotional Quotient (EQ)-Alignment Ability for value-based interactions, and Professional Quotient (PQ)-Professional Expertise for specialized proficiency. For practical value, we pioneer a Value-oriented Evaluation (VQ) framework assessing economic viability, social impact, ethical alignment, and environmental sustainability. Our modular architecture integrates six components with an implementation roadmap. Through analysis of 200+ benchmarks, we identify key challenges including dynamic assessment needs and interpretability gaps. It provides actionable guidance for developing LLMs that are technically proficient, contextually relevant, and ethically sound. We maintain a curated repository of open-source evaluation resources at: https://github.com/onejune2018/Awesome-LLM-Eval.
CandidateDrug4Cancer: An Open Molecular Graph Learning Benchmark on Drug Discovery for Cancer
Mar 02, 2022
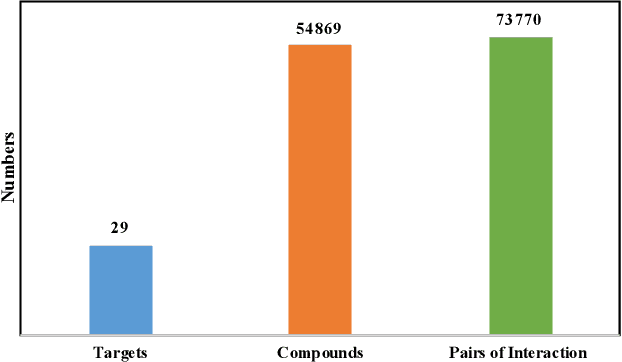
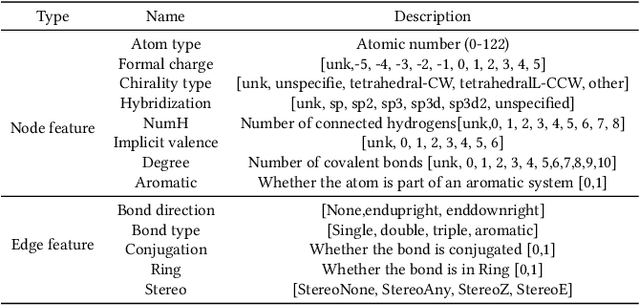
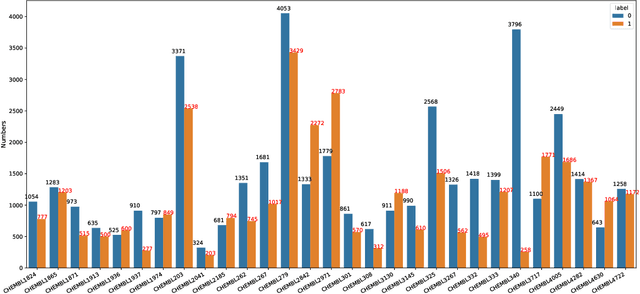
Anti-cancer drug discoveries have been serendipitous, we sought to present the Open Molecular Graph Learning Benchmark, named CandidateDrug4Cancer, a challenging and realistic benchmark dataset to facilitate scalable, robust, and reproducible graph machine learning research for anti-cancer drug discovery. CandidateDrug4Cancer dataset encompasses multiple most-mentioned 29 targets for cancer, covering 54869 cancer-related drug molecules which are ranged from pre-clinical, clinical and FDA-approved. Besides building the datasets, we also perform benchmark experiments with effective Drug Target Interaction (DTI) prediction baselines using descriptors and expressive graph neural networks. Experimental results suggest that CandidateDrug4Cancer presents significant challenges for learning molecular graphs and targets in practical application, indicating opportunities for future researches on developing candidate drugs for treating cancers.
Pairwise Half-graph Discrimination: A Simple Graph-level Self-supervised Strategy for Pre-training Graph Neural Networks
Oct 26, 2021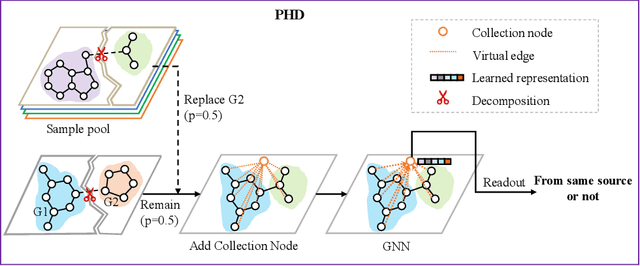
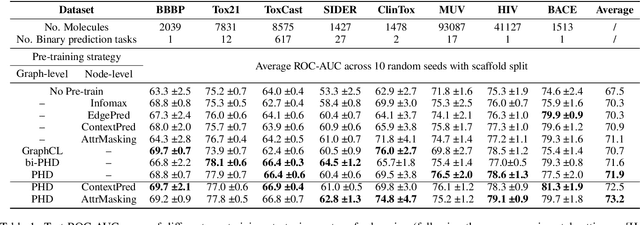
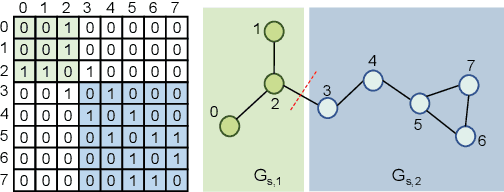
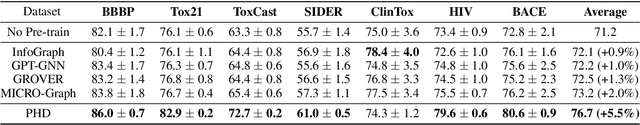
Self-supervised learning has gradually emerged as a powerful technique for graph representation learning. However, transferable, generalizable, and robust representation learning on graph data still remains a challenge for pre-training graph neural networks. In this paper, we propose a simple and effective self-supervised pre-training strategy, named Pairwise Half-graph Discrimination (PHD), that explicitly pre-trains a graph neural network at graph-level. PHD is designed as a simple binary classification task to discriminate whether two half-graphs come from the same source. Experiments demonstrate that the PHD is an effective pre-training strategy that offers comparable or superior performance on 13 graph classification tasks compared with state-of-the-art strategies, and achieves notable improvements when combined with node-level strategies. Moreover, the visualization of learned representation revealed that PHD strategy indeed empowers the model to learn graph-level knowledge like the molecular scaffold. These results have established PHD as a powerful and effective self-supervised learning strategy in graph-level representation learning.
Simulated annealing for optimization of graphs and sequences
Oct 01, 2021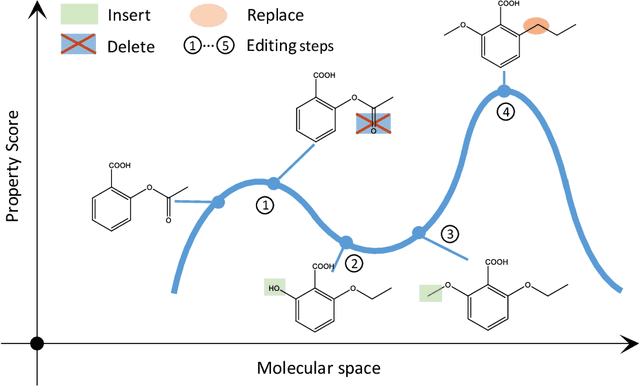
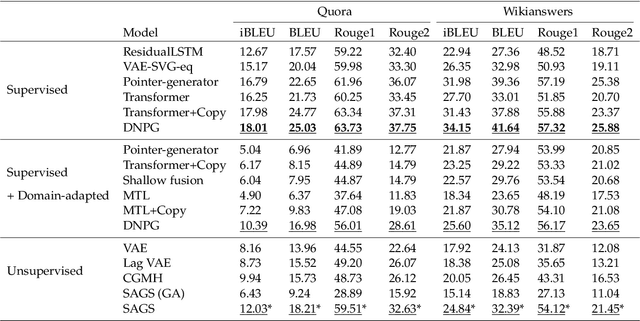
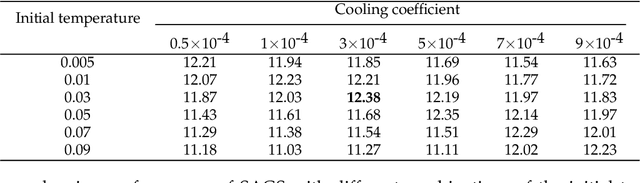
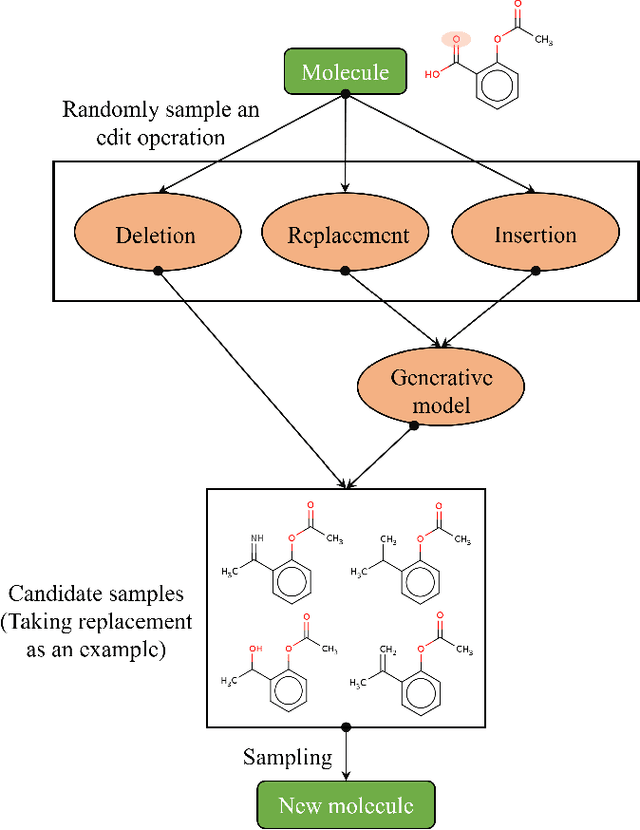
Optimization of discrete structures aims at generating a new structure with the better property given an existing one, which is a fundamental problem in machine learning. Different from the continuous optimization, the realistic applications of discrete optimization (e.g., text generation) are very challenging due to the complex and long-range constraints, including both syntax and semantics, in discrete structures. In this work, we present SAGS, a novel Simulated Annealing framework for Graph and Sequence optimization. The key idea is to integrate powerful neural networks into metaheuristics (e.g., simulated annealing, SA) to restrict the search space in discrete optimization. We start by defining a sophisticated objective function, involving the property of interest and pre-defined constraints (e.g., grammar validity). SAGS searches from the discrete space towards this objective by performing a sequence of local edits, where deep generative neural networks propose the editing content and thus can control the quality of editing. We evaluate SAGS on paraphrase generation and molecule generation for sequence optimization and graph optimization, respectively. Extensive results show that our approach achieves state-of-the-art performance compared with existing paraphrase generation methods in terms of both automatic and human evaluations. Further, SAGS also significantly outperforms all the previous methods in molecule generation.
* This article is an accepted manuscript of Neurocomputing. arXiv admin note: substantial text overlap with arXiv:1909.03588
Learn molecular representations from large-scale unlabeled molecules for drug discovery
Dec 21, 2020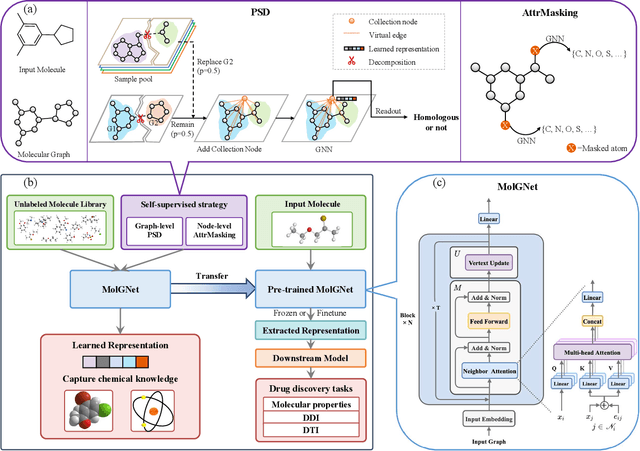
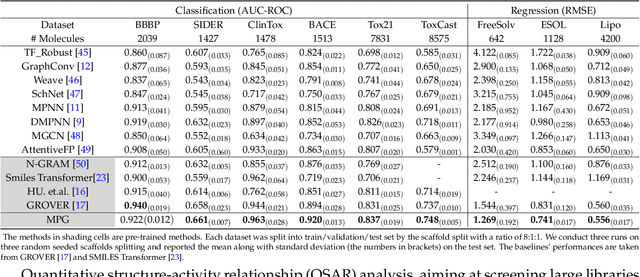
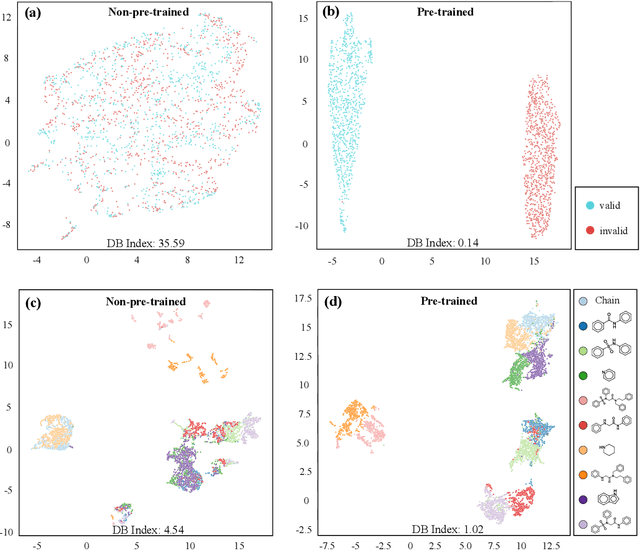
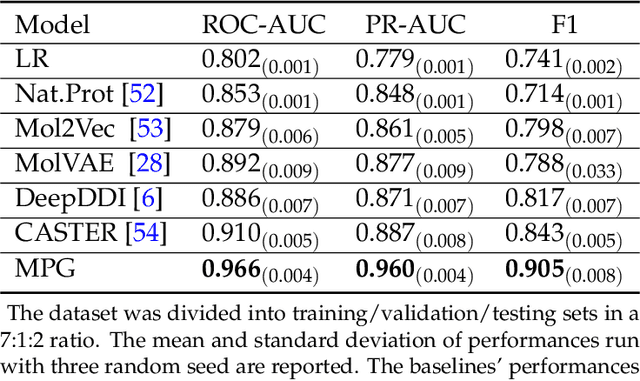
How to produce expressive molecular representations is a fundamental challenge in AI-driven drug discovery. Graph neural network (GNN) has emerged as a powerful technique for modeling molecular data. However, previous supervised approaches usually suffer from the scarcity of labeled data and have poor generalization capability. Here, we proposed a novel Molecular Pre-training Graph-based deep learning framework, named MPG, that leans molecular representations from large-scale unlabeled molecules. In MPG, we proposed a powerful MolGNet model and an effective self-supervised strategy for pre-training the model at both the node and graph-level. After pre-training on 11 million unlabeled molecules, we revealed that MolGNet can capture valuable chemistry insights to produce interpretable representation. The pre-trained MolGNet can be fine-tuned with just one additional output layer to create state-of-the-art models for a wide range of drug discovery tasks, including molecular properties prediction, drug-drug interaction, and drug-target interaction, involving 13 benchmark datasets. Our work demonstrates that MPG is promising to become a novel approach in the drug discovery pipeline.