 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFeatEHR-LLM: Leveraging Large Language Models for Feature Engineering in Electronic Health Records
Apr 24, 2026Feature engineering for Electronic Health Records (EHR) is complicated by irregular observation intervals, variable measurement frequencies, and structural sparsity inherent to clinical time series. Existing automated methods either lack clinical domain awareness or assume clean, regularly sampled inputs, limiting their applicability to real-world EHR data. We present \textbf{FeatEHR-LLM}, a framework that leverages Large Language Models (LLMs) to generate clinically meaningful tabular features from irregularly sampled EHR time series. To limit patient privacy exposure, the LLM operates exclusively on dataset schemas and task descriptions rather than raw patient records. A tool-augmented generation mechanism equips the LLM with specialized routines for querying irregular temporal data, enabling it to produce executable feature-extraction code that explicitly handles uneven observation patterns and informative sparsity. FeatEHR-LLM supports both univariate and multivariate feature generation through an iterative, validation-in-the-loop pipeline. Evaluated on eight clinical prediction tasks across four ICU datasets, our framework achieves the highest mean AUROC on 7 out of 8 tasks, with improvements of up to 6 percentage points over strong baselines. Code is available at github.com/hojjatkarami/FeatEHR-LLM.
ReservoirTTA: Prolonged Test-time Adaptation for Evolving and Recurring Domains
May 20, 2025This paper introduces ReservoirTTA, a novel plug-in framework designed for prolonged test-time adaptation (TTA) in scenarios where the test domain continuously shifts over time, including cases where domains recur or evolve gradually. At its core, ReservoirTTA maintains a reservoir of domain-specialized models -- an adaptive test-time model ensemble -- that both detects new domains via online clustering over style features of incoming samples and routes each sample to the appropriate specialized model, and thereby enables domain-specific adaptation. This multi-model strategy overcomes key limitations of single model adaptation, such as catastrophic forgetting, inter-domain interference, and error accumulation, ensuring robust and stable performance on sustained non-stationary test distributions. Our theoretical analysis reveals key components that bound parameter variance and prevent model collapse, while our plug-in TTA module mitigates catastrophic forgetting of previously encountered domains. Extensive experiments on the classification corruption benchmarks, including ImageNet-C and CIFAR-10/100-C, as well as the Cityscapes$\rightarrow$ACDC semantic segmentation task, covering recurring and continuously evolving domain shifts, demonstrate that ReservoirTTA significantly improves adaptation accuracy and maintains stable performance across prolonged, recurring shifts, outperforming state-of-the-art methods.
Slide-Level Prompt Learning with Vision Language Models for Few-Shot Multiple Instance Learning in Histopathology
Mar 21, 2025In this paper, we address the challenge of few-shot classification in histopathology whole slide images (WSIs) by utilizing foundational vision-language models (VLMs) and slide-level prompt learning. Given the gigapixel scale of WSIs, conventional multiple instance learning (MIL) methods rely on aggregation functions to derive slide-level (bag-level) predictions from patch representations, which require extensive bag-level labels for training. In contrast, VLM-based approaches excel at aligning visual embeddings of patches with candidate class text prompts but lack essential pathological prior knowledge. Our method distinguishes itself by utilizing pathological prior knowledge from language models to identify crucial local tissue types (patches) for WSI classification, integrating this within a VLM-based MIL framework. Our approach effectively aligns patch images with tissue types, and we fine-tune our model via prompt learning using only a few labeled WSIs per category. Experimentation on real-world pathological WSI datasets and ablation studies highlight our method's superior performance over existing MIL- and VLM-based methods in few-shot WSI classification tasks. Our code is publicly available at https://github.com/LTS5/SLIP.
What to align in multimodal contrastive learning?
Sep 11, 2024Humans perceive the world through multisensory integration, blending the information of different modalities to adapt their behavior. Contrastive learning offers an appealing solution for multimodal self-supervised learning. Indeed, by considering each modality as a different view of the same entity, it learns to align features of different modalities in a shared representation space. However, this approach is intrinsically limited as it only learns shared or redundant information between modalities, while multimodal interactions can arise in other ways. In this work, we introduce CoMM, a Contrastive MultiModal learning strategy that enables the communication between modalities in a single multimodal space. Instead of imposing cross- or intra- modality constraints, we propose to align multimodal representations by maximizing the mutual information between augmented versions of these multimodal features. Our theoretical analysis shows that shared, synergistic and unique terms of information naturally emerge from this formulation, allowing us to estimate multimodal interactions beyond redundancy. We test CoMM both in a controlled and in a series of real-world settings: in the former, we demonstrate that CoMM effectively captures redundant, unique and synergistic information between modalities. In the latter, CoMM learns complex multimodal interactions and achieves state-of-the-art results on the six multimodal benchmarks.
Ground-truth effects in learning-based fiber orientation distribution estimation in neonatal brains
Sep 02, 2024



Diffusion Magnetic Resonance Imaging (dMRI) is a non-invasive method for depicting brain microstructure in vivo. Fiber orientation distributions (FODs) are mathematical representations extensively used to map white matter fiber configurations. Recently, FOD estimation with deep neural networks has seen growing success, in particular, those of neonates estimated with fewer diffusion measurements. These methods are mostly trained on target FODs reconstructed with multi-shell multi-tissue constrained spherical deconvolution (MSMT-CSD), which might not be the ideal ground truth for developing brains. Here, we investigate this hypothesis by training a state-of-the-art model based on the U-Net architecture on both MSMT-CSD and single-shell three-tissue constrained spherical deconvolution (SS3T-CSD). Our results suggest that SS3T-CSD might be more suited for neonatal brains, given that the ratio between single and multiple fiber-estimated voxels with SS3T-CSD is more realistic compared to MSMT-CSD. Additionally, increasing the number of input gradient directions significantly improves performance with SS3T-CSD over MSMT-CSD. Finally, in an age domain-shift setting, SS3T-CSD maintains robust performance across age groups, indicating its potential for more accurate neonatal brain imaging.
Spatially Regularized Super-Resolved Constrained Spherical Deconvolution (SR$^2$-CSD) of Diffusion MRI Data
Aug 23, 2024



Constrained Spherical Deconvolution (CSD) is crucial for estimating white matter fiber orientations using diffusion MRI data. A relevant parameter in CSD is the maximum order $l_{max}$ used in the spherical harmonics series, influencing the angular resolution of the Fiber Orientation Distributions (FODs). Lower $l_{max}$ values produce smoother and more stable estimates, but result in reduced angular resolution. Conversely, higher $l_{max}$ values, as employed in the Super-Resolved CSD variant, are essential for resolving narrow inter-fiber angles but lead to spurious lobes due to increased noise sensitivity. To address this issue, we propose a novel Spatially Regularized Super-Resolved CSD (SR$^2$-CSD) approach, incorporating spatial priors into the CSD framework. This method leverages spatial information among adjacent voxels, enhancing the stability and noise robustness of FOD estimations. SR$^2$-CSD facilitates the practical use of Super-Resolved CSD by including a J-invariant auto-calibrated total variation FOD denoiser. We evaluated the performance of SR$^2$-CSD against standard CSD and Super-Resolved CSD using phantom numerical data and various real brain datasets, including a test-retest sample of six subjects scanned twice. In phantom data, SR$^2$-CSD outperformed both CSD and Super-Resolved CSD, reducing the angular error (AE) by approximately half and the peak number error (PNE) by a factor of three across all noise levels considered. In real data, SR$^2$-CSD produced more continuous FOD estimates with higher spatial-angular coherency. In the test-retest sample, SR$^2$-CSD consistently yielded more reproducible estimates, with reduced AE, PNE, mean squared error, and increased angular correlation coefficient between the FODs estimated from the two scans for each subject.
PIV3CAMS: a multi-camera dataset for multiple computer vision problems and its application to novel view-point synthesis
Jul 26, 2024
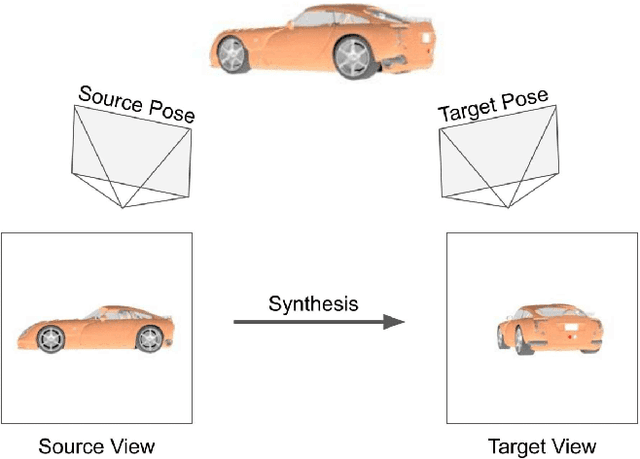


The modern approaches for computer vision tasks significantly rely on machine learning, which requires a large number of quality images. While there is a plethora of image datasets with a single type of images, there is a lack of datasets collected from multiple cameras. In this thesis, we introduce Paired Image and Video data from three CAMeraS, namely PIV3CAMS, aimed at multiple computer vision tasks. The PIV3CAMS dataset consists of 8385 pairs of images and 82 pairs of videos taken from three different cameras: Canon D5 Mark IV, Huawei P20, and ZED stereo camera. The dataset includes various indoor and outdoor scenes from different locations in Zurich (Switzerland) and Cheonan (South Korea). Some of the computer vision applications that can benefit from the PIV3CAMS dataset are image/video enhancement, view interpolation, image matching, and much more. We provide a careful explanation of the data collection process and detailed analysis of the data. The second part of this thesis studies the usage of depth information in the view synthesizing task. In addition to the regeneration of a current state-of-the-art algorithm, we investigate several proposed alternative models that integrate depth information geometrically. Through extensive experiments, we show that the effect of depth is crucial in small view changes. Finally, we apply our model to the introduced PIV3CAMS dataset to synthesize novel target views as an example application of PIV3CAMS.
A Simple Framework for Open-Vocabulary Zero-Shot Segmentation
Jun 23, 2024



Zero-shot classification capabilities naturally arise in models trained within a vision-language contrastive framework. Despite their classification prowess, these models struggle in dense tasks like zero-shot open-vocabulary segmentation. This deficiency is often attributed to the absence of localization cues in captions and the intertwined nature of the learning process, which encompasses both image representation learning and cross-modality alignment. To tackle these issues, we propose SimZSS, a Simple framework for open-vocabulary Zero-Shot Segmentation. The method is founded on two key principles: i) leveraging frozen vision-only models that exhibit spatial awareness while exclusively aligning the text encoder and ii) exploiting the discrete nature of text and linguistic knowledge to pinpoint local concepts within captions. By capitalizing on the quality of the visual representations, our method requires only image-caption pairs datasets and adapts to both small curated and large-scale noisy datasets. When trained on COCO Captions across 8 GPUs, SimZSS achieves state-of-the-art results on 7 out of 8 benchmark datasets in less than 15 minutes.
Uncertainty modeling for fine-tuned implicit functions
Jun 17, 2024



Implicit functions such as Neural Radiance Fields (NeRFs), occupancy networks, and signed distance functions (SDFs) have become pivotal in computer vision for reconstructing detailed object shapes from sparse views. Achieving optimal performance with these models can be challenging due to the extreme sparsity of inputs and distribution shifts induced by data corruptions. To this end, large, noise-free synthetic datasets can serve as shape priors to help models fill in gaps, but the resulting reconstructions must be approached with caution. Uncertainty estimation is crucial for assessing the quality of these reconstructions, particularly in identifying areas where the model is uncertain about the parts it has inferred from the prior. In this paper, we introduce Dropsembles, a novel method for uncertainty estimation in tuned implicit functions. We demonstrate the efficacy of our approach through a series of experiments, starting with toy examples and progressing to a real-world scenario. Specifically, we train a Convolutional Occupancy Network on synthetic anatomical data and test it on low-resolution MRI segmentations of the lumbar spine. Our results show that Dropsembles achieve the accuracy and calibration levels of deep ensembles but with significantly less computational cost.
Un-Mixing Test-Time Normalization Statistics: Combatting Label Temporal Correlation
Jan 16, 2024



In an era where test-time adaptation methods increasingly rely on the nuanced manipulation of batch normalization (BN) parameters, one critical assumption often goes overlooked: that of independently and identically distributed (i.i.d.) test batches with respect to unknown labels. This assumption culminates in biased estimates of BN statistics and jeopardizes system stability under non-i.i.d. conditions. This paper pioneers a departure from the i.i.d. paradigm by introducing a groundbreaking strategy termed "Un-Mixing Test-Time Normalization Statistics" (UnMix-TNS). UnMix-TNS re-calibrates the instance-wise statistics used to normalize each instance in a batch by mixing it with multiple unmixed statistics components, thus inherently simulating the i.i.d. environment. The key lies in our innovative online unmixing procedure, which persistently refines these statistics components by drawing upon the closest instances from an incoming test batch. Remarkably generic in its design, UnMix-TNS seamlessly integrates with an array of state-of-the-art test-time adaptation methods and pre-trained architectures equipped with BN layers. Empirical evaluations corroborate the robustness of UnMix-TNS under varied scenarios ranging from single to continual and mixed domain shifts. UnMix-TNS stands out when handling test data streams with temporal correlation, including those with corrupted real-world non-i.i.d. streams, sustaining its efficacy even with minimal batch sizes and individual samples. Our results set a new standard for test-time adaptation, demonstrating significant improvements in both stability and performance across multiple benchmarks.