 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGeneral Multimodal Protein Design Enables DNA-Encoding of Chemistry
Apr 06, 2026Evolution is an extraordinary engine for enzymatic diversity, yet the chemistry it has explored remains a narrow slice of what DNA can encode. Deep generative models can design new proteins that bind ligands, but none have created enzymes without pre-specifying catalytic residues. We introduce DISCO (DIffusion for Sequence-structure CO-design), a multimodal model that co-designs protein sequence and 3D structure around arbitrary biomolecules, as well as inference-time scaling methods that optimize objectives across both modalities. Conditioned solely on reactive intermediates, DISCO designs diverse heme enzymes with novel active-site geometries. These enzymes catalyze new-to-nature carbene-transfer reactions, including alkene cyclopropanation, spirocyclopropanation, B-H, and C(sp$^3$)-H insertions, with high activities exceeding those of engineered enzymes. Random mutagenesis of a selected design further confirmed that enzyme activity can be improved through directed evolution. By providing a scalable route to evolvable enzymes, DISCO broadens the potential scope of genetically encodable transformations. Code is available at https://github.com/DISCO-design/DISCO.
SynCoGen: Synthesizable 3D Molecule Generation via Joint Reaction and Coordinate Modeling
Jul 16, 2025



Ensuring synthesizability in generative small molecule design remains a major challenge. While recent developments in synthesizable molecule generation have demonstrated promising results, these efforts have been largely confined to 2D molecular graph representations, limiting the ability to perform geometry-based conditional generation. In this work, we present SynCoGen (Synthesizable Co-Generation), a single framework that combines simultaneous masked graph diffusion and flow matching for synthesizable 3D molecule generation. SynCoGen samples from the joint distribution of molecular building blocks, chemical reactions, and atomic coordinates. To train the model, we curated SynSpace, a dataset containing over 600K synthesis-aware building block graphs and 3.3M conformers. SynCoGen achieves state-of-the-art performance in unconditional small molecule graph and conformer generation, and the model delivers competitive performance in zero-shot molecular linker design for protein ligand generation in drug discovery. Overall, this multimodal formulation represents a foundation for future applications enabled by non-autoregressive molecular generation, including analog expansion, lead optimization, and direct structure conditioning.
Generating $π$-Functional Molecules Using STGG+ with Active Learning
Feb 20, 2025Generating novel molecules with out-of-distribution properties is a major challenge in molecular discovery. While supervised learning methods generate high-quality molecules similar to those in a dataset, they struggle to generalize to out-of-distribution properties. Reinforcement learning can explore new chemical spaces but often conducts 'reward-hacking' and generates non-synthesizable molecules. In this work, we address this problem by integrating a state-of-the-art supervised learning method, STGG+, in an active learning loop. Our approach iteratively generates, evaluates, and fine-tunes STGG+ to continuously expand its knowledge. We denote this approach STGG+AL. We apply STGG+AL to the design of organic $\pi$-functional materials, specifically two challenging tasks: 1) generating highly absorptive molecules characterized by high oscillator strength and 2) designing absorptive molecules with reasonable oscillator strength in the near-infrared (NIR) range. The generated molecules are validated and rationalized in-silico with time-dependent density functional theory. Our results demonstrate that our method is highly effective in generating novel molecules with high oscillator strength, contrary to existing methods such as reinforcement learning (RL) methods. We open-source our active-learning code along with our Conjugated-xTB dataset containing 2.9 million $\pi$-conjugated molecules and the function for approximating the oscillator strength and absorption wavelength (based on sTDA-xTB).
Bifurcated Generative Flow Networks
Jun 04, 2024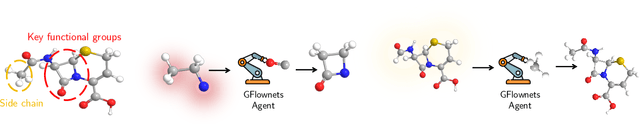

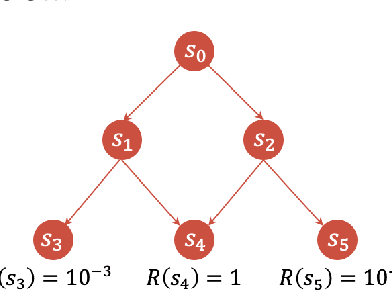
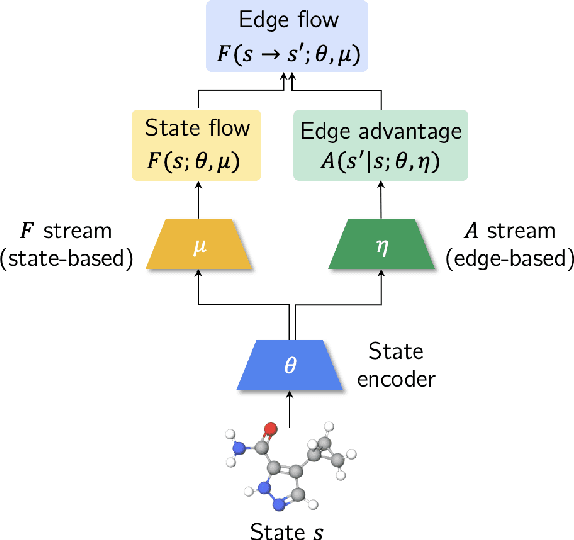
Generative Flow Networks (GFlowNets), a new family of probabilistic samplers, have recently emerged as a promising framework for learning stochastic policies that generate high-quality and diverse objects proportionally to their rewards. However, existing GFlowNets often suffer from low data efficiency due to the direct parameterization of edge flows or reliance on backward policies that may struggle to scale up to large action spaces. In this paper, we introduce Bifurcated GFlowNets (BN), a novel approach that employs a bifurcated architecture to factorize the flows into separate representations for state flows and edge-based flow allocation. This factorization enables BN to learn more efficiently from data and better handle large-scale problems while maintaining the convergence guarantee. Through extensive experiments on standard evaluation benchmarks, we demonstrate that BN significantly improves learning efficiency and effectiveness compared to strong baselines.
Sequence-Augmented SE(3)-Flow Matching For Conditional Protein Backbone Generation
May 30, 2024Proteins are essential for almost all biological processes and derive their diverse functions from complex 3D structures, which are in turn determined by their amino acid sequences. In this paper, we exploit the rich biological inductive bias of amino acid sequences and introduce FoldFlow-2, a novel sequence-conditioned SE(3)-equivariant flow matching model for protein structure generation. FoldFlow-2 presents substantial new architectural features over the previous FoldFlow family of models including a protein large language model to encode sequence, a new multi-modal fusion trunk that combines structure and sequence representations, and a geometric transformer based decoder. To increase diversity and novelty of generated samples -- crucial for de-novo drug design -- we train FoldFlow-2 at scale on a new dataset that is an order of magnitude larger than PDB datasets of prior works, containing both known proteins in PDB and high-quality synthetic structures achieved through filtering. We further demonstrate the ability to align FoldFlow-2 to arbitrary rewards, e.g. increasing secondary structures diversity, by introducing a Reinforced Finetuning (ReFT) objective. We empirically observe that FoldFlow-2 outperforms previous state-of-the-art protein structure-based generative models, improving over RFDiffusion in terms of unconditional generation across all metrics including designability, diversity, and novelty across all protein lengths, as well as exhibiting generalization on the task of equilibrium conformation sampling. Finally, we demonstrate that a fine-tuned FoldFlow-2 makes progress on challenging conditional design tasks such as designing scaffolds for the VHH nanobody.
Generative Active Learning for the Search of Small-molecule Protein Binders
May 02, 2024



Despite substantial progress in machine learning for scientific discovery in recent years, truly de novo design of small molecules which exhibit a property of interest remains a significant challenge. We introduce LambdaZero, a generative active learning approach to search for synthesizable molecules. Powered by deep reinforcement learning, LambdaZero learns to search over the vast space of molecules to discover candidates with a desired property. We apply LambdaZero with molecular docking to design novel small molecules that inhibit the enzyme soluble Epoxide Hydrolase 2 (sEH), while enforcing constraints on synthesizability and drug-likeliness. LambdaZero provides an exponential speedup in terms of the number of calls to the expensive molecular docking oracle, and LambdaZero de novo designed molecules reach docking scores that would otherwise require the virtual screening of a hundred billion molecules. Importantly, LambdaZero discovers novel scaffolds of synthesizable, drug-like inhibitors for sEH. In in vitro experimental validation, a series of ligands from a generated quinazoline-based scaffold were synthesized, and the lead inhibitor N-(4,6-di(pyrrolidin-1-yl)quinazolin-2-yl)-N-methylbenzamide (UM0152893) displayed sub-micromolar enzyme inhibition of sEH.
Iterated Denoising Energy Matching for Sampling from Boltzmann Densities
Feb 09, 2024



Efficiently generating statistically independent samples from an unnormalized probability distribution, such as equilibrium samples of many-body systems, is a foundational problem in science. In this paper, we propose Iterated Denoising Energy Matching (iDEM), an iterative algorithm that uses a novel stochastic score matching objective leveraging solely the energy function and its gradient -- and no data samples -- to train a diffusion-based sampler. Specifically, iDEM alternates between (I) sampling regions of high model density from a diffusion-based sampler and (II) using these samples in our stochastic matching objective to further improve the sampler. iDEM is scalable to high dimensions as the inner matching objective, is simulation-free, and requires no MCMC samples. Moreover, by leveraging the fast mode mixing behavior of diffusion, iDEM smooths out the energy landscape enabling efficient exploration and learning of an amortized sampler. We evaluate iDEM on a suite of tasks ranging from standard synthetic energy functions to invariant $n$-body particle systems. We show that the proposed approach achieves state-of-the-art performance on all metrics and trains $2-5\times$ faster, which allows it to be the first method to train using energy on the challenging $55$-particle Lennard-Jones system.
Towards equilibrium molecular conformation generation with GFlowNets
Oct 20, 2023



Sampling diverse, thermodynamically feasible molecular conformations plays a crucial role in predicting properties of a molecule. In this paper we propose to use GFlowNet for sampling conformations of small molecules from the Boltzmann distribution, as determined by the molecule's energy. The proposed approach can be used in combination with energy estimation methods of different fidelity and discovers a diverse set of low-energy conformations for highly flexible drug-like molecules. We demonstrate that GFlowNet can reproduce molecular potential energy surfaces by sampling proportionally to the Boltzmann distribution.
Diffusion Generative Flow Samplers: Improving learning signals through partial trajectory optimization
Oct 04, 2023



We tackle the problem of sampling from intractable high-dimensional density functions, a fundamental task that often appears in machine learning and statistics. We extend recent sampling-based approaches that leverage controlled stochastic processes to model approximate samples from these target densities. The main drawback of these approaches is that the training objective requires full trajectories to compute, resulting in sluggish credit assignment issues due to use of entire trajectories and a learning signal present only at the terminal time. In this work, we present Diffusion Generative Flow Samplers (DGFS), a sampling-based framework where the learning process can be tractably broken down into short partial trajectory segments, via parameterizing an additional "flow function". Our method takes inspiration from the theory developed for generative flow networks (GFlowNets), allowing us to make use of intermediate learning signals and benefit from off-policy exploration capabilities. Through a variety of challenging experiments, we demonstrate that DGFS results in more accurate estimates of the normalization constant than closely-related prior methods.
SE(3)-Stochastic Flow Matching for Protein Backbone Generation
Oct 03, 2023The computational design of novel protein structures has the potential to impact numerous scientific disciplines greatly. Toward this goal, we introduce $\text{FoldFlow}$ a series of novel generative models of increasing modeling power based on the flow-matching paradigm over $3\text{D}$ rigid motions -- i.e. the group $\text{SE(3)}$ -- enabling accurate modeling of protein backbones. We first introduce $\text{FoldFlow-Base}$, a simulation-free approach to learning deterministic continuous-time dynamics and matching invariant target distributions on $\text{SE(3)}$. We next accelerate training by incorporating Riemannian optimal transport to create $\text{FoldFlow-OT}$, leading to the construction of both more simple and stable flows. Finally, we design $\text{FoldFlow-SFM}$ coupling both Riemannian OT and simulation-free training to learn stochastic continuous-time dynamics over $\text{SE(3)}$. Our family of $\text{FoldFlow}$ generative models offer several key advantages over previous approaches to the generative modeling of proteins: they are more stable and faster to train than diffusion-based approaches, and our models enjoy the ability to map any invariant source distribution to any invariant target distribution over $\text{SE(3)}$. Empirically, we validate our FoldFlow models on protein backbone generation of up to $300$ amino acids leading to high-quality designable, diverse, and novel samples.