 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePRiMeFlow: Capturing Complex Expression Heterogeneity in Perturbation Response Modelling
Apr 15, 2026Predicting the effects of perturbations in-silico on cell state can identify drivers of cell behavior at scale and accelerate drug discovery. However, modeling challenges remain due to the inherent heterogeneity of single cell gene expression and the complex, latent gene dependencies. Here, we present PRiMeFlow, an end-to-end flow matching based approach to directly model the effects of genetic and small molecule perturbations in the gene expression space. The distribution-fitting approach taken by PRiMeFlow enables it to accurately approximate the empirical distribution of single-cell gene expression, which we demonstrate through extensive benchmarking inside PerturBench. Through ablation studies, we also validate important model design choices such as operating in gene expression space and parameterizing the velocity field with a U-Net architecture. The PRiMeFlow architecture was used as the basis for the model that won the Generalist Prize in the first ARC Virtual Cell Challenge.
Towards equilibrium molecular conformation generation with GFlowNets
Oct 20, 2023



Sampling diverse, thermodynamically feasible molecular conformations plays a crucial role in predicting properties of a molecule. In this paper we propose to use GFlowNet for sampling conformations of small molecules from the Boltzmann distribution, as determined by the molecule's energy. The proposed approach can be used in combination with energy estimation methods of different fidelity and discovers a diverse set of low-energy conformations for highly flexible drug-like molecules. We demonstrate that GFlowNet can reproduce molecular potential energy surfaces by sampling proportionally to the Boltzmann distribution.
PhyloGFN: Phylogenetic inference with generative flow networks
Oct 12, 2023Phylogenetics is a branch of computational biology that studies the evolutionary relationships among biological entities. Its long history and numerous applications notwithstanding, inference of phylogenetic trees from sequence data remains challenging: the high complexity of tree space poses a significant obstacle for the current combinatorial and probabilistic techniques. In this paper, we adopt the framework of generative flow networks (GFlowNets) to tackle two core problems in phylogenetics: parsimony-based and Bayesian phylogenetic inference. Because GFlowNets are well-suited for sampling complex combinatorial structures, they are a natural choice for exploring and sampling from the multimodal posterior distribution over tree topologies and evolutionary distances. We demonstrate that our amortized posterior sampler, PhyloGFN, produces diverse and high-quality evolutionary hypotheses on real benchmark datasets. PhyloGFN is competitive with prior works in marginal likelihood estimation and achieves a closer fit to the target distribution than state-of-the-art variational inference methods.
Neural representation and generation for RNA secondary structures
Feb 01, 2021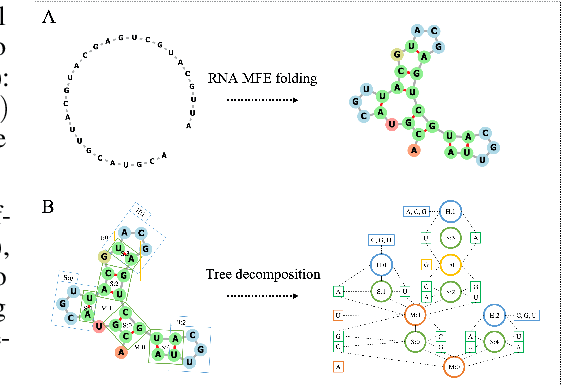
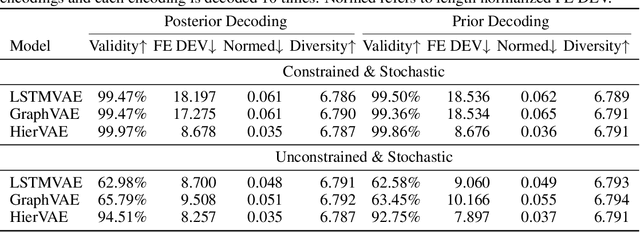
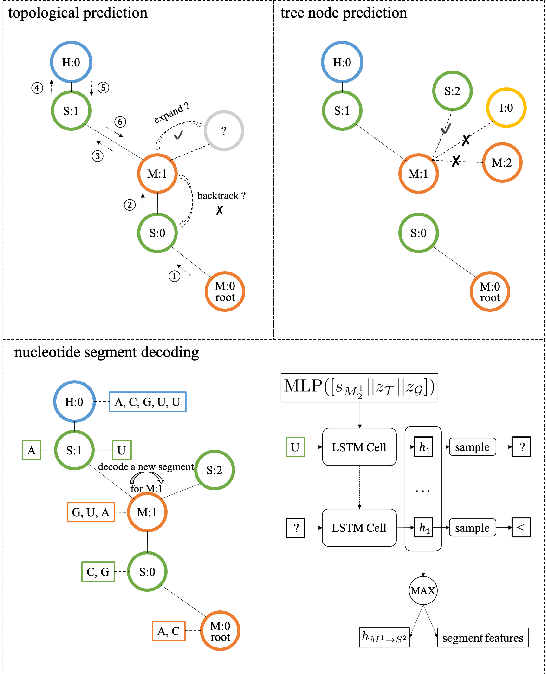
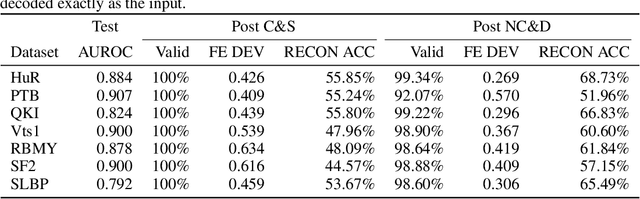
Our work is concerned with the generation and targeted design of RNA, a type of genetic macromolecule that can adopt complex structures which influence their cellular activities and functions. The design of large scale and complex biological structures spurs dedicated graph-based deep generative modeling techniques, which represents a key but underappreciated aspect of computational drug discovery. In this work, we investigate the principles behind representing and generating different RNA structural modalities, and propose a flexible framework to jointly embed and generate these molecular structures along with their sequence in a meaningful latent space. Equipped with a deep understanding of RNA molecular structures, our most sophisticated encoding and decoding methods operate on the molecular graph as well as the junction tree hierarchy, integrating strong inductive bias about RNA structural regularity and folding mechanism such that high structural validity, stability and diversity of generated RNAs are achieved. Also, we seek to adequately organize the latent space of RNA molecular embeddings with regard to the interaction with proteins, and targeted optimization is used to navigate in this latent space to search for desired novel RNA molecules.