 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAn Equivariant Generative Framework for Molecular Graph-Structure Co-Design
Apr 12, 2023Designing molecules with desirable physiochemical properties and functionalities is a long-standing challenge in chemistry, material science, and drug discovery. Recently, machine learning-based generative models have emerged as promising approaches for \emph{de novo} molecule design. However, further refinement of methodology is highly desired as most existing methods lack unified modeling of 2D topology and 3D geometry information and fail to effectively learn the structure-property relationship for molecule design. Here we present MolCode, a roto-translation equivariant generative framework for \underline{Mol}ecular graph-structure \underline{Co-de}sign. In MolCode, 3D geometric information empowers the molecular 2D graph generation, which in turn helps guide the prediction of molecular 3D structure. Extensive experimental results show that MolCode outperforms previous methods on a series of challenging tasks including \emph{de novo} molecule design, targeted molecule discovery, and structure-based drug design. Particularly, MolCode not only consistently generates valid (99.95$\%$ Validity) and diverse (98.75$\%$ Uniqueness) molecular graphs/structures with desirable properties, but also generate drug-like molecules with high affinity to target proteins (61.8$\%$ high-affinity ratio), which demonstrates MolCode's potential applications in material design and drug discovery. Our extensive investigation reveals that the 2D topology and 3D geometry contain intrinsically complementary information in molecule design, and provide new insights into machine learning-based molecule representation and generation.
Hierarchical Graph Transformer with Adaptive Node Sampling
Oct 08, 2022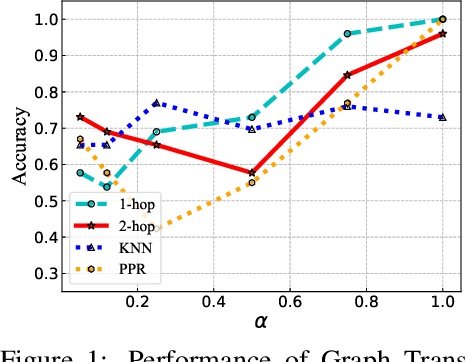
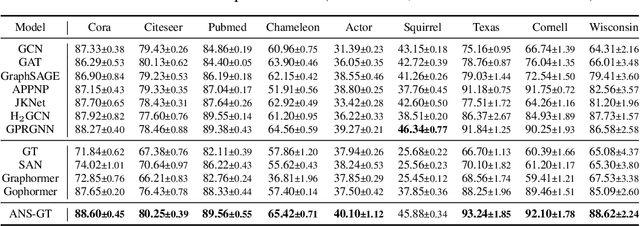
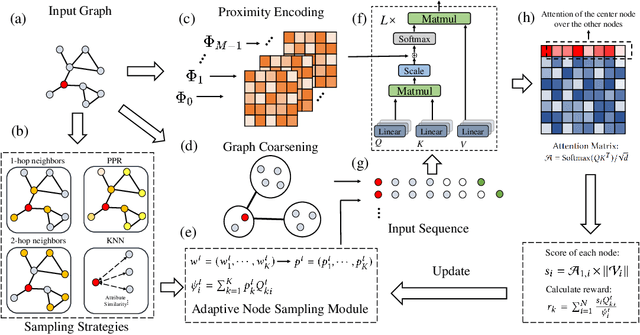
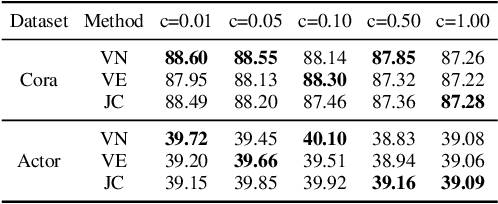
The Transformer architecture has achieved remarkable success in a number of domains including natural language processing and computer vision. However, when it comes to graph-structured data, transformers have not achieved competitive performance, especially on large graphs. In this paper, we identify the main deficiencies of current graph transformers:(1) Existing node sampling strategies in Graph Transformers are agnostic to the graph characteristics and the training process. (2) Most sampling strategies only focus on local neighbors and neglect the long-range dependencies in the graph. We conduct experimental investigations on synthetic datasets to show that existing sampling strategies are sub-optimal. To tackle the aforementioned problems, we formulate the optimization strategies of node sampling in Graph Transformer as an adversary bandit problem, where the rewards are related to the attention weights and can vary in the training procedure. Meanwhile, we propose a hierarchical attention scheme with graph coarsening to capture the long-range interactions while reducing computational complexity. Finally, we conduct extensive experiments on real-world datasets to demonstrate the superiority of our method over existing graph transformers and popular GNNs.
Model Inversion Attacks against Graph Neural Networks
Sep 19, 2022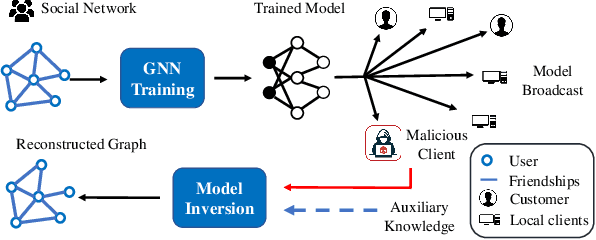
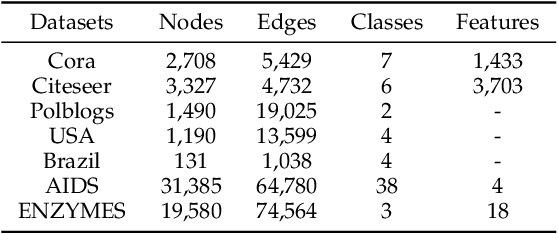
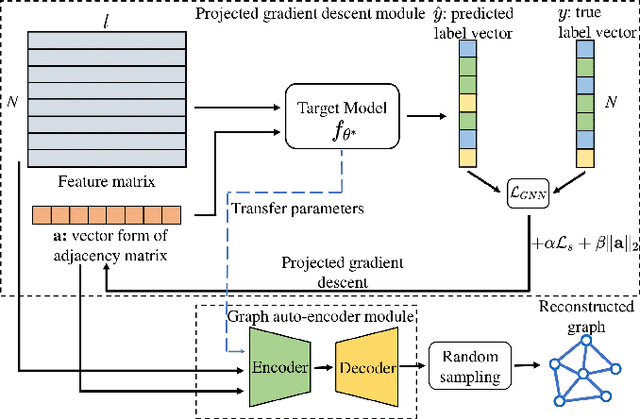
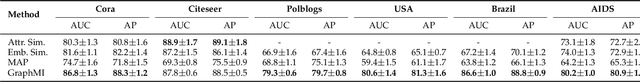
Many data mining tasks rely on graphs to model relational structures among individuals (nodes). Since relational data are often sensitive, there is an urgent need to evaluate the privacy risks in graph data. One famous privacy attack against data analysis models is the model inversion attack, which aims to infer sensitive data in the training dataset and leads to great privacy concerns. Despite its success in grid-like domains, directly applying model inversion attacks on non-grid domains such as graph leads to poor attack performance. This is mainly due to the failure to consider the unique properties of graphs. To bridge this gap, we conduct a systematic study on model inversion attacks against Graph Neural Networks (GNNs), one of the state-of-the-art graph analysis tools in this paper. Firstly, in the white-box setting where the attacker has full access to the target GNN model, we present GraphMI to infer the private training graph data. Specifically, in GraphMI, a projected gradient module is proposed to tackle the discreteness of graph edges and preserve the sparsity and smoothness of graph features; a graph auto-encoder module is used to efficiently exploit graph topology, node attributes, and target model parameters for edge inference; a random sampling module can finally sample discrete edges. Furthermore, in the hard-label black-box setting where the attacker can only query the GNN API and receive the classification results, we propose two methods based on gradient estimation and reinforcement learning (RL-GraphMI). Our experimental results show that such defenses are not sufficiently effective and call for more advanced defenses against privacy attacks.
A photonic chip-based machine learning approach for the prediction of molecular properties
Mar 03, 2022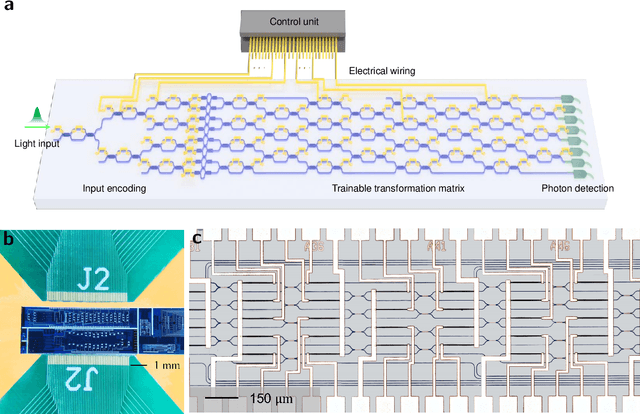
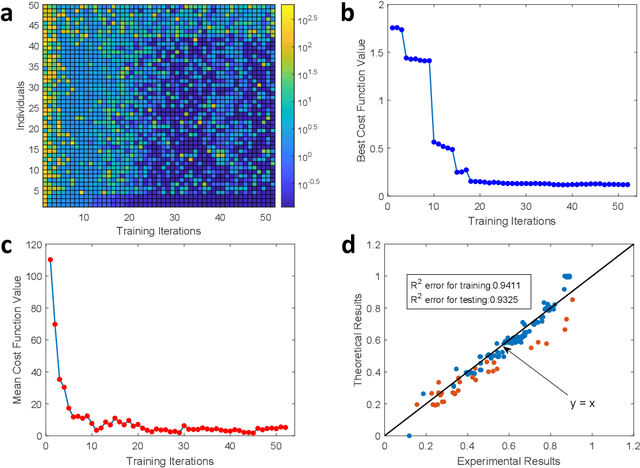
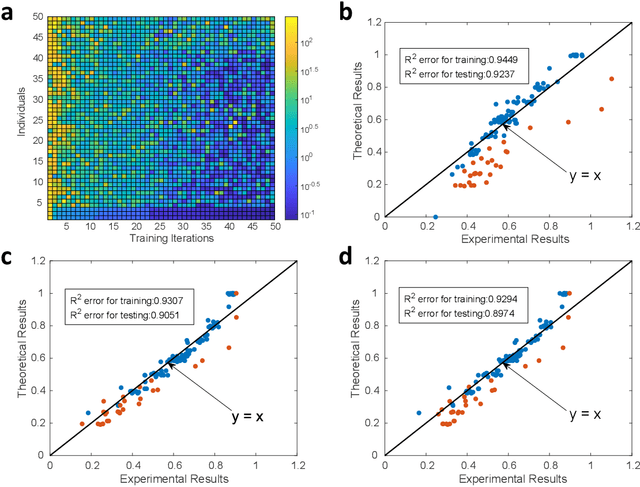
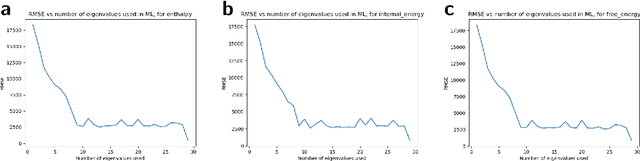
Machine learning methods have revolutionized the discovery process of new molecules and materials. However, the intensive training process of neural networks for molecules with ever increasing complexity has resulted in exponential growth in computation cost, leading to long simulation time and high energy consumption. Photonic chip technology offers an alternative platform for implementing neural network with faster data processing and lower energy usage compared to digital computers. Here, we demonstrate the capability of photonic neural networks in predicting the quantum mechanical properties of molecules. Additionally, we show that multiple properties can be learned simultaneously in a photonic chip via a multi-task regression learning algorithm, which we believe is the first of its kind, as most previous works focus on implementing a network for the task of classification. Photonics technology are also naturally capable of implementing complex-valued neural networks at no additional hardware cost and we show that such neural networks outperform conventional real-valued networks for molecular property prediction. Our work opens the avenue for harnessing photonic technology for large-scale machine learning applications in molecular sciences such as drug discovery and materials design.
Motif-based Graph Self-Supervised Learning for Molecular Property Prediction
Oct 16, 2021
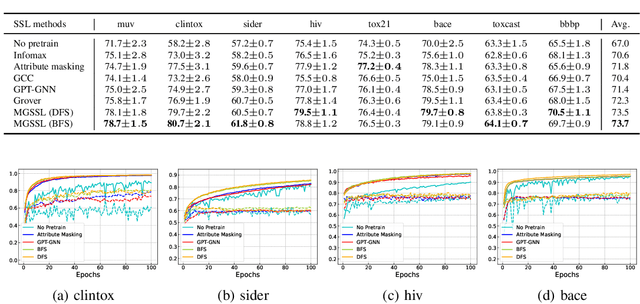
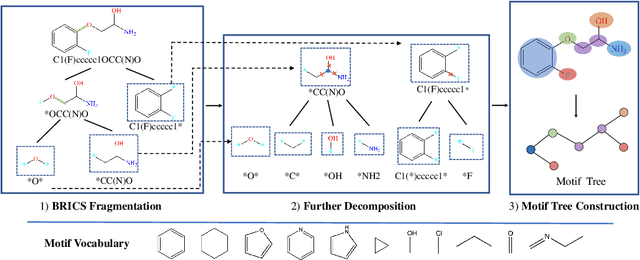
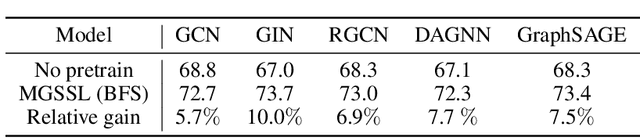
Predicting molecular properties with data-driven methods has drawn much attention in recent years. Particularly, Graph Neural Networks (GNNs) have demonstrated remarkable success in various molecular generation and prediction tasks. In cases where labeled data is scarce, GNNs can be pre-trained on unlabeled molecular data to first learn the general semantic and structural information before being fine-tuned for specific tasks. However, most existing self-supervised pre-training frameworks for GNNs only focus on node-level or graph-level tasks. These approaches cannot capture the rich information in subgraphs or graph motifs. For example, functional groups (frequently-occurred subgraphs in molecular graphs) often carry indicative information about the molecular properties. To bridge this gap, we propose Motif-based Graph Self-supervised Learning (MGSSL) by introducing a novel self-supervised motif generation framework for GNNs. First, for motif extraction from molecular graphs, we design a molecule fragmentation method that leverages a retrosynthesis-based algorithm BRICS and additional rules for controlling the size of motif vocabulary. Second, we design a general motif-based generative pre-training framework in which GNNs are asked to make topological and label predictions. This generative framework can be implemented in two different ways, i.e., breadth-first or depth-first. Finally, to take the multi-scale information in molecular graphs into consideration, we introduce a multi-level self-supervised pre-training. Extensive experiments on various downstream benchmark tasks show that our methods outperform all state-of-the-art baselines.
Alchemy: A Quantum Chemistry Dataset for Benchmarking AI Models
Jun 22, 2019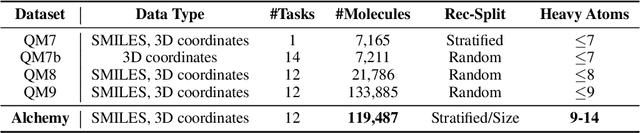
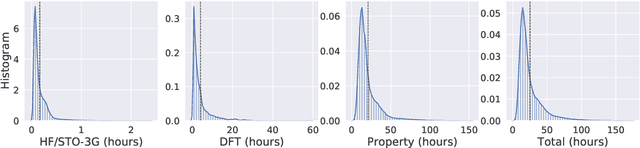
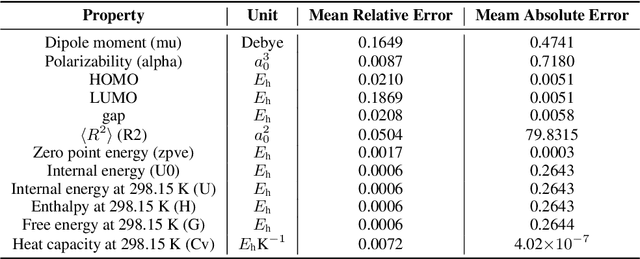
We introduce a new molecular dataset, named Alchemy, for developing machine learning models useful in chemistry and material science. As of June 20th 2019, the dataset comprises of 12 quantum mechanical properties of 119,487 organic molecules with up to 14 heavy atoms, sampled from the GDB MedChem database. The Alchemy dataset expands the volume and diversity of existing molecular datasets. Our extensive benchmarks of the state-of-the-art graph neural network models on Alchemy clearly manifest the usefulness of new data in validating and developing machine learning models for chemistry and material science. We further launch a contest to attract attentions from researchers in the related fields. More details can be found on the contest website \footnote{https://alchemy.tencent.com}. At the time of benchamrking experiment, we have generated 119,487 molecules in our Alchemy dataset. More molecular samples are generated since then. Hence, we provide a list of molecules used in the reported benchmarks.