 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeScalable Spatio-Temporal SE(3) Diffusion for Long-Horizon Protein Dynamics
Feb 02, 2026Molecular dynamics (MD) simulations remain the gold standard for studying protein dynamics, but their computational cost limits access to biologically relevant timescales. Recent generative models have shown promise in accelerating simulations, yet they struggle with long-horizon generation due to architectural constraints, error accumulation, and inadequate modeling of spatio-temporal dynamics. We present STAR-MD (Spatio-Temporal Autoregressive Rollout for Molecular Dynamics), a scalable SE(3)-equivariant diffusion model that generates physically plausible protein trajectories over microsecond timescales. Our key innovation is a causal diffusion transformer with joint spatio-temporal attention that efficiently captures complex space-time dependencies while avoiding the memory bottlenecks of existing methods. On the standard ATLAS benchmark, STAR-MD achieves state-of-the-art performance across all metrics--substantially improving conformational coverage, structural validity, and dynamic fidelity compared to previous methods. STAR-MD successfully extrapolates to generate stable microsecond-scale trajectories where baseline methods fail catastrophically, maintaining high structural quality throughout the extended rollout. Our comprehensive evaluation reveals severe limitations in current models for long-horizon generation, while demonstrating that STAR-MD's joint spatio-temporal modeling enables robust dynamics simulation at biologically relevant timescales, paving the way for accelerated exploration of protein function.
MatterTune: An Integrated, User-Friendly Platform for Fine-Tuning Atomistic Foundation Models to Accelerate Materials Simulation and Discovery
Apr 14, 2025



Geometric machine learning models such as graph neural networks have achieved remarkable success in recent years in chemical and materials science research for applications such as high-throughput virtual screening and atomistic simulations. The success of these models can be attributed to their ability to effectively learn latent representations of atomic structures directly from the training data. Conversely, this also results in high data requirements for these models, hindering their application to problems which are data sparse which are common in this domain. To address this limitation, there is a growing development in the area of pre-trained machine learning models which have learned general, fundamental, geometric relationships in atomistic data, and which can then be fine-tuned to much smaller application-specific datasets. In particular, models which are pre-trained on diverse, large-scale atomistic datasets have shown impressive generalizability and flexibility to downstream applications, and are increasingly referred to as atomistic foundation models. To leverage the untapped potential of these foundation models, we introduce MatterTune, a modular and extensible framework that provides advanced fine-tuning capabilities and seamless integration of atomistic foundation models into downstream materials informatics and simulation workflows, thereby lowering the barriers to adoption and facilitating diverse applications in materials science. In its current state, MatterTune supports a number of state-of-the-art foundation models such as ORB, MatterSim, JMP, and EquformerV2, and hosts a wide range of features including a modular and flexible design, distributed and customizable fine-tuning, broad support for downstream informatics tasks, and more.
Distribution Learning for Molecular Regression
Jul 30, 2024



Using "soft" targets to improve model performance has been shown to be effective in classification settings, but the usage of soft targets for regression is a much less studied topic in machine learning. The existing literature on the usage of soft targets for regression fails to properly assess the method's limitations, and empirical evaluation is quite limited. In this work, we assess the strengths and drawbacks of existing methods when applied to molecular property regression tasks. Our assessment outlines key biases present in existing methods and proposes methods to address them, evaluated through careful ablation studies. We leverage these insights to propose Distributional Mixture of Experts (DMoE): A model-independent, and data-independent method for regression which trains a model to predict probability distributions of its targets. Our proposed loss function combines the cross entropy between predicted and target distributions and the L1 distance between their expected values to produce a loss function that is robust to the outlined biases. We evaluate the performance of DMoE on different molecular property prediction datasets -- Open Catalyst (OC20), MD17, and QM9 -- across different backbone model architectures -- SchNet, GemNet, and Graphormer. Our results demonstrate that the proposed method is a promising alternative to classical regression for molecular property prediction tasks, showing improvements over baselines on all datasets and architectures.
From Molecules to Materials: Pre-training Large Generalizable Models for Atomic Property Prediction
Oct 25, 2023Foundation models have been transformational in machine learning fields such as natural language processing and computer vision. Similar success in atomic property prediction has been limited due to the challenges of training effective models across multiple chemical domains. To address this, we introduce Joint Multi-domain Pre-training (JMP), a supervised pre-training strategy that simultaneously trains on multiple datasets from different chemical domains, treating each dataset as a unique pre-training task within a multi-task framework. Our combined training dataset consists of $\sim$120M systems from OC20, OC22, ANI-1x, and Transition-1x. We evaluate performance and generalization by fine-tuning over a diverse set of downstream tasks and datasets including: QM9, rMD17, MatBench, QMOF, SPICE, and MD22. JMP demonstrates an average improvement of 59% over training from scratch, and matches or sets state-of-the-art on 34 out of 40 tasks. Our work highlights the potential of pre-training strategies that utilize diverse data to advance property prediction across chemical domains, especially for low-data tasks.
The Open Catalyst 2022 Dataset and Challenges for Oxide Electrocatalysis
Jun 17, 2022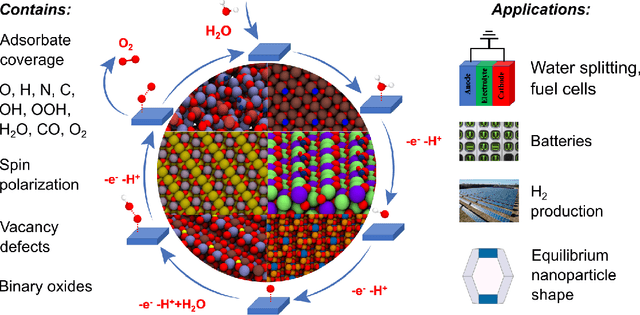
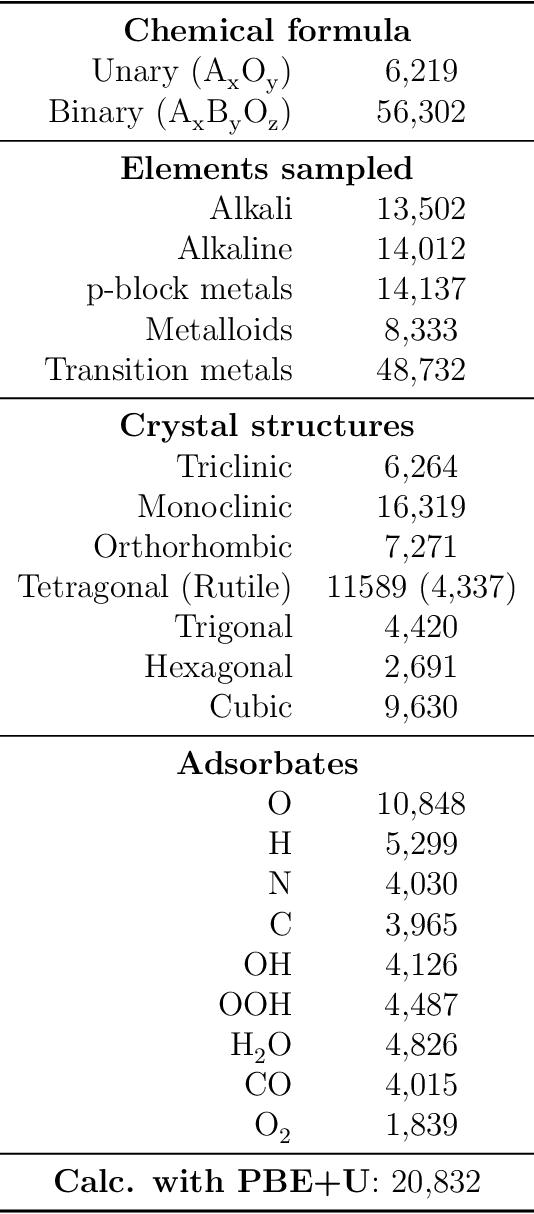
Computational catalysis and machine learning communities have made considerable progress in developing machine learning models for catalyst discovery and design. Yet, a general machine learning potential that spans the chemical space of catalysis is still out of reach. A significant hurdle is obtaining access to training data across a wide range of materials. One important class of materials where data is lacking are oxides, which inhibits models from studying the Oxygen Evolution Reaction and oxide electrocatalysis more generally. To address this we developed the Open Catalyst 2022(OC22) dataset, consisting of 62,521 Density Functional Theory (DFT) relaxations (~9,884,504 single point calculations) across a range of oxide materials, coverages, and adsorbates (*H, *O, *N, *C, *OOH, *OH, *OH2, *O2, *CO). We define generalized tasks to predict the total system energy that are applicable across catalysis, develop baseline performance of several graph neural networks (SchNet, DimeNet++, ForceNet, SpinConv, PaiNN, GemNet-dT, GemNet-OC), and provide pre-defined dataset splits to establish clear benchmarks for future efforts. For all tasks, we study whether combining datasets leads to better results, even if they contain different materials or adsorbates. Specifically, we jointly train models on Open Catalyst 2020 (OC20) Dataset and OC22, or fine-tune pretrained OC20 models on OC22. In the most general task, GemNet-OC sees a ~32% improvement in energy predictions through fine-tuning and a ~9% improvement in force predictions via joint training. Surprisingly, joint training on both the OC20 and much smaller OC22 datasets also improves total energy predictions on OC20 by ~19%. The dataset and baseline models are open sourced, and a public leaderboard will follow to encourage continued community developments on the total energy tasks and data.