 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMORI-Seg: Learning Morphological Geometry for Instance Segmentation without Instance Annotations
May 27, 2026Instance-level quantification of kidney functional units is essential for morphometric analysis, yet most publicly available pathology datasets provide only semantic segmentation annotations, where adjacent structures of the same class are merged into single regions. This prevents reliable instance-level analysis and limits downstream quantitative studies. Existing heuristic post-processing methods often yield suboptimal instance separation, particularly in crowded and adherent regions, while deep learning-based instance segmentation approaches typically require intensive instance-level annotations that are costly and labor-intensive to obtain. We propose MORI-Seg, a deep learning framework that enables instance segmentation without requiring instance-level annotations. Instead of heuristic splitting or instance supervision, MORI-Seg learns morphology-aware geometric representations directly from semantic masks by jointly modeling object-centric distance fields and boundary-band representations to encode interior structure and contact interfaces. A class-conditioned feature disentanglement module further promotes intra-instance coherence and inter-instance separation. Under semantic-only supervision, MORI-Seg decomposes connected semantic regions into distinct instance masks in an end-to-end manner. Experiments demonstrate improved instance separation accuracy and more reliable morphometric quantification compared with classical post-processing pipelines and representative semantic-to-instance learning approaches. The official implementation is publicly available at https://github.com/ddrrnn123/MORI-Seg.
DUET: Dual-Paradigm Adaptive Expert Triage with Single-cell Inductive Prior for Spatial Transcriptomics Prediction
May 13, 2026Inferring spatially resolved gene expression from histology images offers a cost-effective complement to spatial transcriptomics (ST). However, existing methods reduce this task to a simple morphology-to-expression mapping, where visual similarity does not guarantee molecular consistency. Meanwhile, single-cell data has amassed rich resources far surpassing the scale of ST data, yet it remains underexplored in vision-omics modeling. Furthermore, current approaches commit to a monolithic paradigm with bottlenecks, unable to balance expressive flexibility with biological fidelity. To bridge these gaps, we propose DUET, a novel dual-paradigm framework that synergizes parametric prediction and memory-based retrieval under cellular inductive priors. DUET implements a parallel regression-retrieval paradigm, adaptively reconciling the outputs of its complementary pathways. To mitigate aleatoric vision ambiguity, we incorporate large-scale single-cell references to impose molecular states as biological constraints for faithful learning. Building upon structural refinement, we further design a lightweight adapter to dynamically assign branch preference across spatial contexts to achieve optimal performance. Extensive experiments on three public datasets across varied gene scales demonstrate that DUET achieves SOTA performance, with consistent gains contributed by each proposed component. Code is available at https://github.com/Junchao-Zhu/DUET
Explainable Pathomics Feature Visualization via Correlation-aware Conditional Feature Editing
Feb 05, 2026Pathomics is a recent approach that offers rich quantitative features beyond what black-box deep learning can provide, supporting more reproducible and explainable biomarkers in digital pathology. However, many derived features (e.g., "second-order moment") remain difficult to interpret, especially across different clinical contexts, which limits their practical adoption. Conditional diffusion models show promise for explainability through feature editing, but they typically assume feature independence**--**an assumption violated by intrinsically correlated pathomics features. Consequently, editing one feature while fixing others can push the model off the biological manifold and produce unrealistic artifacts. To address this, we propose a Manifold-Aware Diffusion (MAD) framework for controllable and biologically plausible cell nuclei editing. Unlike existing approaches, our method regularizes feature trajectories within a disentangled latent space learned by a variational auto-encoder (VAE). This ensures that manipulating a target feature automatically adjusts correlated attributes to remain within the learned distribution of real cells. These optimized features then guide a conditional diffusion model to synthesize high-fidelity images. Experiments demonstrate that our approach is able to navigate the manifold of pathomics features when editing those features. The proposed method outperforms baseline methods in conditional feature editing while preserving structural coherence.
MASC: Metal-Aware Sampling and Correction via Reinforcement Learning for Accelerated MRI
Jan 30, 2026Metal implants in MRI cause severe artifacts that degrade image quality and hinder clinical diagnosis. Traditional approaches address metal artifact reduction (MAR) and accelerated MRI acquisition as separate problems. We propose MASC, a unified reinforcement learning framework that jointly optimizes metal-aware k-space sampling and artifact correction for accelerated MRI. To enable supervised training, we construct a paired MRI dataset using physics-based simulation, generating k-space data and reconstructions for phantoms with and without metal implants. This paired dataset provides simulated 3D MRI scans with and without metal implants, where each metal-corrupted sample has an exactly matched clean reference, enabling direct supervision for both artifact reduction and acquisition policy learning. We formulate active MRI acquisition as a sequential decision-making problem, where an artifact-aware Proximal Policy Optimization (PPO) agent learns to select k-space phase-encoding lines under a limited acquisition budget. The agent operates on undersampled reconstructions processed through a U-Net-based MAR network, learning patterns that maximize reconstruction quality. We further propose an end-to-end training scheme where the acquisition policy learns to select k-space lines that best support artifact removal while the MAR network simultaneously adapts to the resulting undersampling patterns. Experiments demonstrate that MASC's learned policies outperform conventional sampling strategies, and end-to-end training improves performance compared to using a frozen pre-trained MAR network, validating the benefit of joint optimization. Cross-dataset experiments on FastMRI with physics-based artifact simulation further confirm generalization to realistic clinical MRI data. The code and models of MASC have been made publicly available: https://github.com/hrlblab/masc
AdaFuse: Adaptive Multimodal Fusion for Lung Cancer Risk Prediction via Reinforcement Learning
Jan 30, 2026Multimodal fusion has emerged as a promising paradigm for disease diagnosis and prognosis, integrating complementary information from heterogeneous data sources such as medical images, clinical records, and radiology reports. However, existing fusion methods process all available modalities through the network, either treating them equally or learning to assign different contribution weights, leaving a fundamental question unaddressed: for a given patient, should certain modalities be used at all? We present AdaFuse, an adaptive multimodal fusion framework that leverages reinforcement learning (RL) to learn patient-specific modality selection and fusion strategies for lung cancer risk prediction. AdaFuse formulates multimodal fusion as a sequential decision process, where the policy network iteratively decides whether to incorporate an additional modality or proceed to prediction based on the information already acquired. This sequential formulation enables the model to condition each selection on previously observed modalities and terminate early when sufficient information is available, rather than committing to a fixed subset upfront. We evaluate AdaFuse on the National Lung Screening Trial (NLST) dataset. Experimental results demonstrate that AdaFuse achieves the highest AUC (0.762) compared to the best single-modality baseline (0.732), the best fixed fusion strategy (0.759), and adaptive baselines including DynMM (0.754) and MoE (0.742), while using fewer FLOPs than all triple-modality methods. Our work demonstrates the potential of reinforcement learning for personalized multimodal fusion in medical imaging, representing a shift from uniform fusion strategies toward adaptive diagnostic pipelines that learn when to consult additional modalities and when existing information suffices for accurate prediction.
HistoWAS: A Pathomics Framework for Large-Scale Feature-Wide Association Studies of Tissue Topology and Patient Outcomes
Dec 23, 2025High-throughput "pathomic" analysis of Whole Slide Images (WSIs) offers new opportunities to study tissue characteristics and for biomarker discovery. However, the clinical relevance of the tissue characteristics at the micro- and macro-environment level is limited by the lack of tools that facilitate the measurement of the spatial interaction of individual structure characteristics and their association with clinical parameters. To address these challenges, we introduce HistoWAS (Histology-Wide Association Study), a computational framework designed to link tissue spatial organization to clinical outcomes. Specifically, HistoWAS implements (1) a feature space that augments conventional metrics with 30 topological and spatial features, adapted from Geographic Information Systems (GIS) point pattern analysis, to quantify tissue micro-architecture; and (2) an association study engine, inspired by Phenome-Wide Association Studies (PheWAS), that performs mass univariate regression for each feature with statistical correction. As a proof of concept, we applied HistoWAS to analyze a total of 102 features (72 conventional object-level features and our 30 spatial features) using 385 PAS-stained WSIs from 206 participants in the Kidney Precision Medicine Project (KPMP). The code and data have been released to https://github.com/hrlblab/histoWAS.
SCR2-ST: Combine Single Cell with Spatial Transcriptomics for Efficient Active Sampling via Reinforcement Learning
Dec 15, 2025



Spatial transcriptomics (ST) is an emerging technology that enables researchers to investigate the molecular relationships underlying tissue morphology. However, acquiring ST data remains prohibitively expensive, and traditional fixed-grid sampling strategies lead to redundant measurements of morphologically similar or biologically uninformative regions, thus resulting in scarce data that constrain current methods. The well-established single-cell sequencing field, however, could provide rich biological data as an effective auxiliary source to mitigate this limitation. To bridge these gaps, we introduce SCR2-ST, a unified framework that leverages single-cell prior knowledge to guide efficient data acquisition and accurate expression prediction. SCR2-ST integrates a single-cell guided reinforcement learning-based (SCRL) active sampling and a hybrid regression-retrieval prediction network SCR2Net. SCRL combines single-cell foundation model embeddings with spatial density information to construct biologically grounded reward signals, enabling selective acquisition of informative tissue regions under constrained sequencing budgets. SCR2Net then leverages the actively sampled data through a hybrid architecture combining regression-based modeling with retrieval-augmented inference, where a majority cell-type filtering mechanism suppresses noisy matches and retrieved expression profiles serve as soft labels for auxiliary supervision. We evaluated SCR2-ST on three public ST datasets, demonstrating SOTA performance in both sampling efficiency and prediction accuracy, particularly under low-budget scenarios. Code is publicly available at: https://github.com/hrlblab/SCR2ST
How Close Are We? Limitations and Progress of AI Models in Banff Lesion Scoring
Oct 31, 2025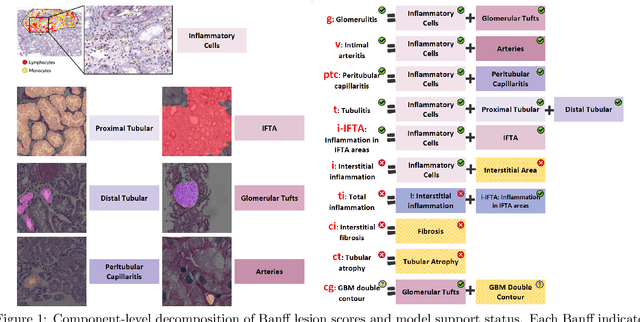
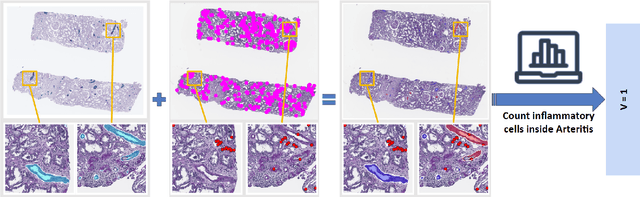
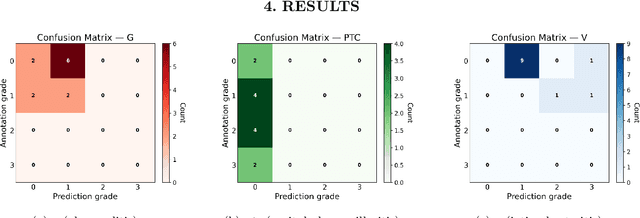
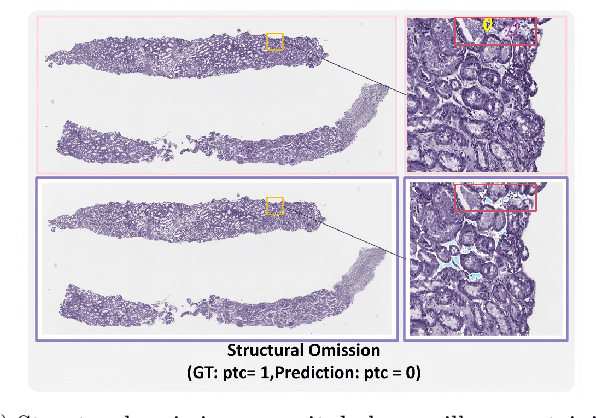
The Banff Classification provides the global standard for evaluating renal transplant biopsies, yet its semi-quantitative nature, complex criteria, and inter-observer variability present significant challenges for computational replication. In this study, we explore the feasibility of approximating Banff lesion scores using existing deep learning models through a modular, rule-based framework. We decompose each Banff indicator - such as glomerulitis (g), peritubular capillaritis (ptc), and intimal arteritis (v) - into its constituent structural and inflammatory components, and assess whether current segmentation and detection tools can support their computation. Model outputs are mapped to Banff scores using heuristic rules aligned with expert guidelines, and evaluated against expert-annotated ground truths. Our findings highlight both partial successes and critical failure modes, including structural omission, hallucination, and detection ambiguity. Even when final scores match expert annotations, inconsistencies in intermediate representations often undermine interpretability. These results reveal the limitations of current AI pipelines in replicating computational expert-level grading, and emphasize the importance of modular evaluation and computational Banff grading standard in guiding future model development for transplant pathology.
DyMorph-B2I: Dynamic and Morphology-Guided Binary-to-Instance Segmentation for Renal Pathology
Aug 21, 2025Accurate morphological quantification of renal pathology functional units relies on instance-level segmentation, yet most existing datasets and automated methods provide only binary (semantic) masks, limiting the precision of downstream analyses. Although classical post-processing techniques such as watershed, morphological operations, and skeletonization, are often used to separate semantic masks into instances, their individual effectiveness is constrained by the diverse morphologies and complex connectivity found in renal tissue. In this study, we present DyMorph-B2I, a dynamic, morphology-guided binary-to-instance segmentation pipeline tailored for renal pathology. Our approach integrates watershed, skeletonization, and morphological operations within a unified framework, complemented by adaptive geometric refinement and customizable hyperparameter tuning for each class of functional unit. Through systematic parameter optimization, DyMorph-B2I robustly separates adherent and heterogeneous structures present in binary masks. Experimental results demonstrate that our method outperforms individual classical approaches and na\"ive combinations, enabling superior instance separation and facilitating more accurate morphometric analysis in renal pathology workflows. The pipeline is publicly available at: https://github.com/ddrrnn123/DyMorph-B2I.
Scale-up Unlearnable Examples Learning with High-Performance Computing
Jan 10, 2025



Recent advancements in AI models are structured to retain user interactions, which could inadvertently include sensitive healthcare data. In the healthcare field, particularly when radiologists use AI-driven diagnostic tools hosted on online platforms, there is a risk that medical imaging data may be repurposed for future AI training without explicit consent, spotlighting critical privacy and intellectual property concerns around healthcare data usage. Addressing these privacy challenges, a novel approach known as Unlearnable Examples (UEs) has been introduced, aiming to make data unlearnable to deep learning models. A prominent method within this area, called Unlearnable Clustering (UC), has shown improved UE performance with larger batch sizes but was previously limited by computational resources. To push the boundaries of UE performance with theoretically unlimited resources, we scaled up UC learning across various datasets using Distributed Data Parallel (DDP) training on the Summit supercomputer. Our goal was to examine UE efficacy at high-performance computing (HPC) levels to prevent unauthorized learning and enhance data security, particularly exploring the impact of batch size on UE's unlearnability. Utilizing the robust computational capabilities of the Summit, extensive experiments were conducted on diverse datasets such as Pets, MedMNist, Flowers, and Flowers102. Our findings reveal that both overly large and overly small batch sizes can lead to performance instability and affect accuracy. However, the relationship between batch size and unlearnability varied across datasets, highlighting the necessity for tailored batch size strategies to achieve optimal data protection. Our results underscore the critical role of selecting appropriate batch sizes based on the specific characteristics of each dataset to prevent learning and ensure data security in deep learning applications.