 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeRing-lite: Scalable Reasoning via C3PO-Stabilized Reinforcement Learning for LLMs
Jun 18, 2025We present Ring-lite, a Mixture-of-Experts (MoE)-based large language model optimized via reinforcement learning (RL) to achieve efficient and robust reasoning capabilities. Built upon the publicly available Ling-lite model, a 16.8 billion parameter model with 2.75 billion activated parameters, our approach matches the performance of state-of-the-art (SOTA) small-scale reasoning models on challenging benchmarks (e.g., AIME, LiveCodeBench, GPQA-Diamond) while activating only one-third of the parameters required by comparable models. To accomplish this, we introduce a joint training pipeline integrating distillation with RL, revealing undocumented challenges in MoE RL training. First, we identify optimization instability during RL training, and we propose Constrained Contextual Computation Policy Optimization(C3PO), a novel approach that enhances training stability and improves computational throughput via algorithm-system co-design methodology. Second, we empirically demonstrate that selecting distillation checkpoints based on entropy loss for RL training, rather than validation metrics, yields superior performance-efficiency trade-offs in subsequent RL training. Finally, we develop a two-stage training paradigm to harmonize multi-domain data integration, addressing domain conflicts that arise in training with mixed dataset. We will release the model, dataset, and code.
The Graph Convolutional Network with Multi-representation Alignment for Drug Synergy Prediction
Nov 27, 2023



Drug combination refers to the use of two or more drugs to treat a specific disease at the same time. It is currently the mainstream way to treat complex diseases. Compared with single drugs, drug combinations have better efficacy and can better inhibit toxicity and drug resistance. The computational model based on deep learning concatenates the representation of multiple drugs and the corresponding cell line feature as input, and the output is whether the drug combination can have an inhibitory effect on the cell line. However, this strategy of concatenating multiple representations has the following defects: the alignment of drug representation and cell line representation is ignored, resulting in the synergistic relationship not being reflected positionally in the embedding space. Moreover, the alignment measurement function in deep learning cannot be suitable for drug synergy prediction tasks due to differences in input types. Therefore, in this work, we propose a graph convolutional network with multi-representation alignment (GCNMRA) for predicting drug synergy. In the GCNMRA model, we designed a multi-representation alignment function suitable for the drug synergy prediction task so that the positional relationship between drug representations and cell line representation is reflected in the embedding space. In addition, the vector modulus of drug representations and cell line representation is considered to improve the accuracy of calculation results and accelerate model convergence. Finally, many relevant experiments were run on multiple drug synergy datasets to verify the effectiveness of the above innovative elements and the excellence of the GCNMRA model.
GraphCL-DTA: a graph contrastive learning with molecular semantics for drug-target binding affinity prediction
Jul 18, 2023



Drug-target binding affinity prediction plays an important role in the early stages of drug discovery, which can infer the strength of interactions between new drugs and new targets. However, the performance of previous computational models is limited by the following drawbacks. The learning of drug representation relies only on supervised data, without taking into account the information contained in the molecular graph itself. Moreover, most previous studies tended to design complicated representation learning module, while uniformity, which is used to measure representation quality, is ignored. In this study, we propose GraphCL-DTA, a graph contrastive learning with molecular semantics for drug-target binding affinity prediction. In GraphCL-DTA, we design a graph contrastive learning framework for molecular graphs to learn drug representations, so that the semantics of molecular graphs are preserved. Through this graph contrastive framework, a more essential and effective drug representation can be learned without additional supervised data. Next, we design a new loss function that can be directly used to smoothly adjust the uniformity of drug and target representations. By directly optimizing the uniformity of representations, the representation quality of drugs and targets can be improved. The effectiveness of the above innovative elements is verified on two real datasets, KIBA and Davis. The excellent performance of GraphCL-DTA on the above datasets suggests its superiority to the state-of-the-art model.
ALT: An Automatic System for Long Tail Scenario Modeling
May 19, 2023



In this paper, we consider the problem of long tail scenario modeling with budget limitation, i.e., insufficient human resources for model training stage and limited time and computing resources for model inference stage. This problem is widely encountered in various applications, yet has received deficient attention so far. We present an automatic system named ALT to deal with this problem. Several efforts are taken to improve the algorithms used in our system, such as employing various automatic machine learning related techniques, adopting the meta learning philosophy, and proposing an essential budget-limited neural architecture search method, etc. Moreover, to build the system, many optimizations are performed from a systematic perspective, and essential modules are armed, making the system more feasible and efficient. We perform abundant experiments to validate the effectiveness of our system and demonstrate the usefulness of the critical modules in our system. Moreover, online results are provided, which fully verified the efficacy of our system.
DCMT: A Direct Entire-Space Causal Multi-Task Framework for Post-Click Conversion Estimation
Feb 13, 2023



In recommendation scenarios, there are two long-standing challenges, i.e., selection bias and data sparsity, which lead to a significant drop in prediction accuracy for both Click-Through Rate (CTR) and post-click Conversion Rate (CVR) tasks. To cope with these issues, existing works emphasize on leveraging Multi-Task Learning (MTL) frameworks (Category 1) or causal debiasing frameworks (Category 2) to incorporate more auxiliary data in the entire exposure/inference space D or debias the selection bias in the click/training space O. However, these two kinds of solutions cannot effectively address the not-missing-at-random problem and debias the selection bias in O to fit the inference in D. To fill the research gaps, we propose a Direct entire-space Causal Multi-Task framework, namely DCMT, for post-click conversion prediction in this paper. Specifically, inspired by users' decision process of conversion, we propose a new counterfactual mechanism to debias the selection bias in D, which can predict the factual CVR and the counterfactual CVR under the soft constraint of a counterfactual prior knowledge. Extensive experiments demonstrate that our DCMT can improve the state-of-the-art methods by an average of 1.07% in terms of CVR AUC on the five offline datasets and 0.75% in terms of PV-CVR on the online A/B test (the Alipay Search). Such improvements can increase millions of conversions per week in real industrial applications, e.g., the Alipay Search.
Self-supervised Learning for Label Sparsity in Computational Drug Repositioning
Jun 01, 2022

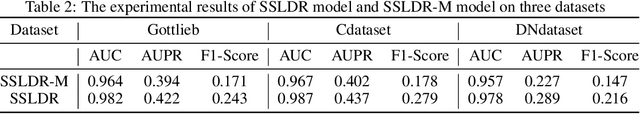
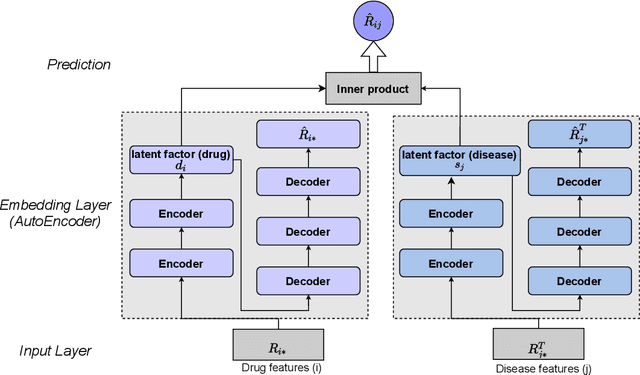
The computational drug repositioning aims to discover new uses for marketed drugs, which can accelerate the drug development process and play an important role in the existing drug discovery system. However, the number of validated drug-disease associations is scarce compared to the number of drugs and diseases in the real world. Too few labeled samples will make the classification model unable to learn effective latent factors of drugs, resulting in poor generalization performance. In this work, we propose a multi-task self-supervised learning framework for computational drug repositioning. The framework tackles label sparsity by learning a better drug representation. Specifically, we take the drug-disease association prediction problem as the main task, and the auxiliary task is to use data augmentation strategies and contrast learning to mine the internal relationships of the original drug features, so as to automatically learn a better drug representation without supervised labels. And through joint training, it is ensured that the auxiliary task can improve the prediction accuracy of the main task. More precisely, the auxiliary task improves drug representation and serving as additional regularization to improve generalization. Furthermore, we design a multi-input decoding network to improve the reconstruction ability of the autoencoder model. We evaluate our model using three real-world datasets. The experimental results demonstrate the effectiveness of the multi-task self-supervised learning framework, and its predictive ability is superior to the state-of-the-art model.
The Computational Drug Repositioning without Negative Sampling
Nov 29, 2021
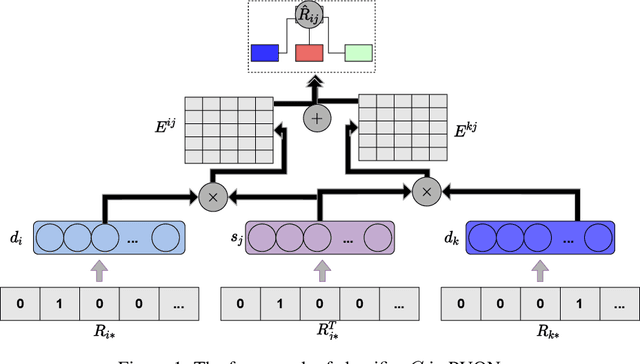
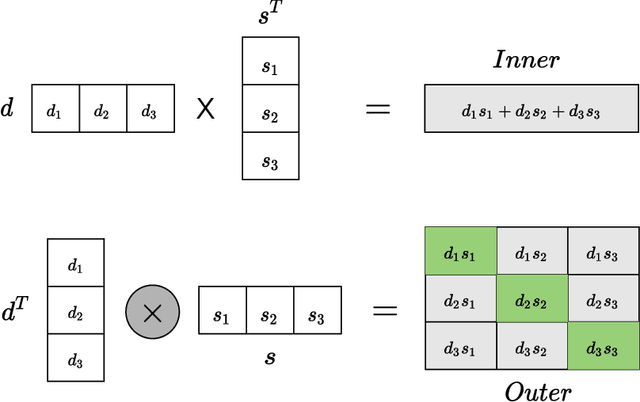
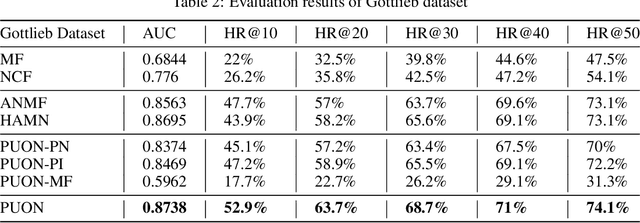
Computational drug repositioning technology is an effective tool to accelerate drug development. Although this technique has been widely used and successful in recent decades, many existing models still suffer from multiple drawbacks such as the massive number of unvalidated drug-disease associations and inner product in the matrix factorization model. The limitations of these works are mainly due to the following two reasons: first, previous works used negative sampling techniques to treat unvalidated drug-disease associations as negative samples, which is invalid in real-world settings; Second, the inner product lacks modeling on the crossover information between dimensions of the latent factor. In this paper, we propose a novel PUON framework for addressing the above deficiencies, which models the joint distribution of drug-disease associations using validated and unvalidated drug-disease associations without employing negative sampling techniques. The PUON also modeled the cross-information of the latent factor of drugs and diseases using the outer product operation. For a comprehensive comparison, we considered 7 popular baselines. Extensive experiments in two real-world datasets showed that PUON achieved the best performance based on 6 popular evaluation metrics.
The Neural Metric Factorization for Computational Drug Repositioning
Oct 08, 2021
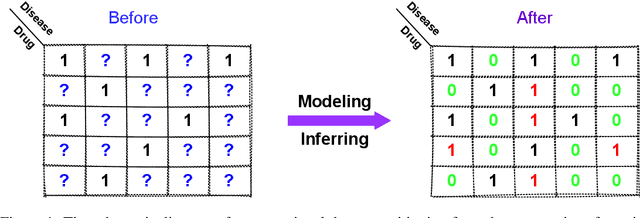
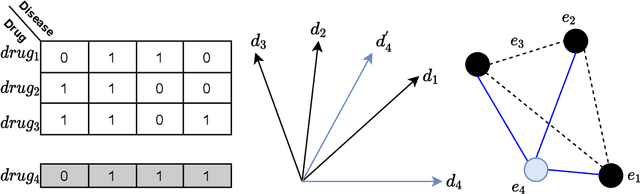
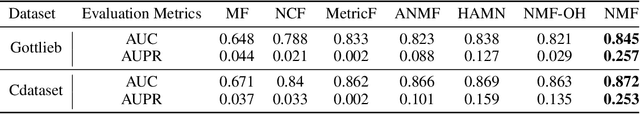
Computational drug repositioning aims to discover new therapeutic diseases for marketed drugs and has the advantages of low cost, short development cycle, and high controllability compared to traditional drug development. The matrix factorization model has become the cornerstone technique for computational drug repositioning due to its ease of implementation and excellent scalability. However, the matrix factorization model uses the inner product operation to represent the association between drugs and diseases, which is lacking in expressive ability. Moreover, the degree of similarity of drugs or diseases could not be implied on their respective latent factor vectors, which is not satisfy the common sense of drug discovery. Therefore, a neural metric factorization model (NMF) for computational drug repositioning is proposed in this work. We novelly consider the latent factor vector of drugs and diseases as a point in the high-dimensional coordinate system and propose a generalized Euclidean distance to represent the association between drugs and diseases to compensate for the shortcomings of the inner product operation. Furthermore, by embedding multiple drug (disease) metrics information into the encoding space of the latent factor vector, the information about the similarity between drugs (diseases) can be reflected in the distance between latent factor vectors. Finally, we conduct wide analysis experiments on two real datasets to demonstrate the effectiveness of the above improvement points and the superiority of the NMF model.
Hybrid Attentional Memory Network for Computational drug repositioning
Jun 12, 2020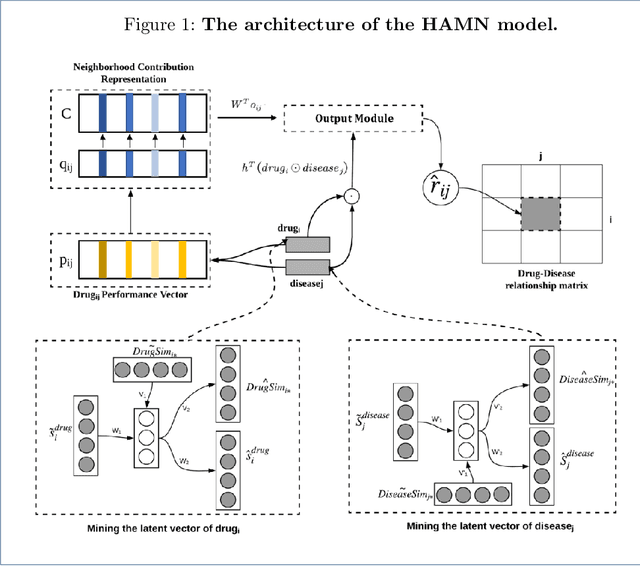

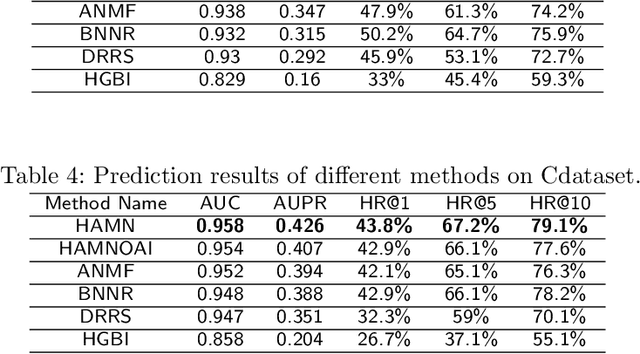
Drug repositioning is designed to discover new uses of known drugs, which is an important and efficient method of drug discovery. Researchers only use one certain type of Collaborative Filtering (CF) models for drug repositioning currently, like the neighborhood based approaches which are good at mining the local information contained in few strong drug-disease associations, or the latent factor based models which are effectively capture the global information shared by a majority of drug-disease associations. Few researchers have combined these two types of CF models to derive a hybrid model with the advantages of both of them. Besides, the cold start problem has always been a major challenge in the field of computational drug repositioning, which restricts the inference ability of relevant models. Inspired by the memory network, we propose the Hybrid Attentional Memory Network (HAMN) model, a deep architecture combines two classes of CF model in a nonlinear manner. Firstly, the memory unit and the attention mechanism are combined to generate the neighborhood contribution representation to capture the local structure of few strong drug-disease associations. Then a variant version of the autoencoder is used to extract the latent factor of drugs and diseases to capture the overall information shared by a majority of drug-disease associations. In that process, ancillary information of drugs and diseases can help to alleviate the cold start problem. Finally, in the prediction stage, the neighborhood contribution representation is combined with the drug latent factor and disease latent factor to produce the predicted value. Comprehensive experimental results on two real data sets show that our proposed HAMN model is superior to other comparison models according to the AUC, AUPR and HR indicators.
InfDetect: a Large Scale Graph-based Fraud Detection System for E-Commerce Insurance
Mar 12, 2020
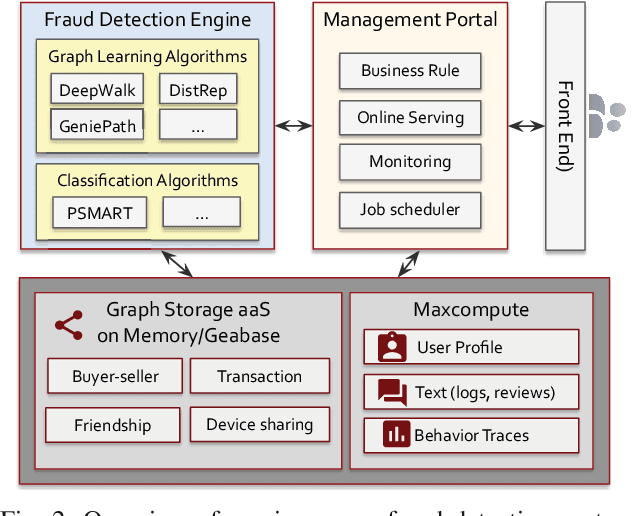
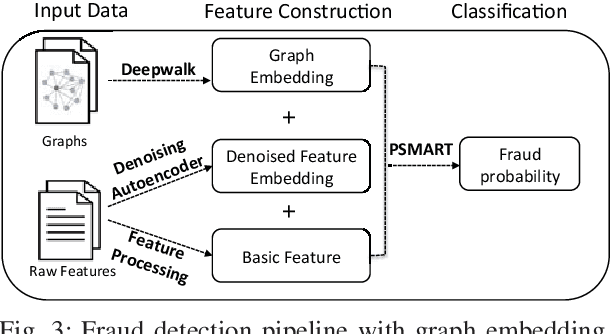
The insurance industry has been creating innovative products around the emerging online shopping activities. Such e-commerce insurance is designed to protect buyers from potential risks such as impulse purchases and counterfeits. Fraudulent claims towards online insurance typically involve multiple parties such as buyers, sellers, and express companies, and they could lead to heavy financial losses. In order to uncover the relations behind organized fraudsters and detect fraudulent claims, we developed a large-scale insurance fraud detection system, i.e., InfDetect, which provides interfaces for commonly used graphs, standard data processing procedures, and a uniform graph learning platform. InfDetect is able to process big graphs containing up to 100 millions of nodes and billions of edges. In this paper, we investigate different graphs to facilitate fraudster mining, such as a device-sharing graph, a transaction graph, a friendship graph, and a buyer-seller graph. These graphs are fed to a uniform graph learning platform containing supervised and unsupervised graph learning algorithms. Cases on widely applied e-commerce insurance are described to demonstrate the usage and capability of our system. InfDetect has successfully detected thousands of fraudulent claims and saved over tens of thousands of dollars daily.