 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAlphaFold Distillation for Improved Inverse Protein Folding
Oct 05, 2022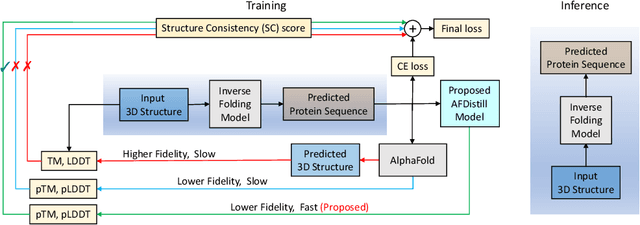
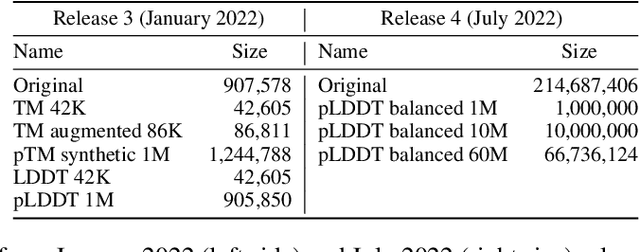
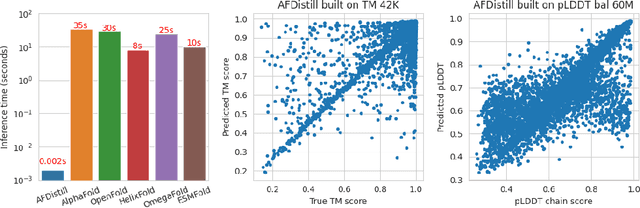

Inverse protein folding, i.e., designing sequences that fold into a given three-dimensional structure, is one of the fundamental design challenges in bio-engineering and drug discovery. Traditionally, inverse folding mainly involves learning from sequences that have an experimentally resolved structure. However, the known structures cover only a tiny space of the protein sequences, imposing limitations on the model learning. Recently proposed forward folding models, e.g., AlphaFold, offer unprecedented opportunity for accurate estimation of the structure given a protein sequence. Naturally, incorporating a forward folding model as a component of an inverse folding approach offers the potential of significantly improving the inverse folding, as the folding model can provide a feedback on any generated sequence in the form of the predicted protein structure or a structural confidence metric. However, at present, these forward folding models are still prohibitively slow to be a part of the model optimization loop during training. In this work, we propose to perform knowledge distillation on the folding model's confidence metrics, e.g., pTM or pLDDT scores, to obtain a smaller, faster and end-to-end differentiable distilled model, which then can be included as part of the structure consistency regularized inverse folding model training. Moreover, our regularization technique is general enough and can be applied in other design tasks, e.g., sequence-based protein infilling. Extensive experiments show a clear benefit of our method over the non-regularized baselines. For example, in inverse folding design problems we observe up to 3% improvement in sequence recovery and up to 45% improvement in protein diversity, while still preserving structural consistency of the generated sequences.
Cloud-Based Real-Time Molecular Screening Platform with MolFormer
Aug 13, 2022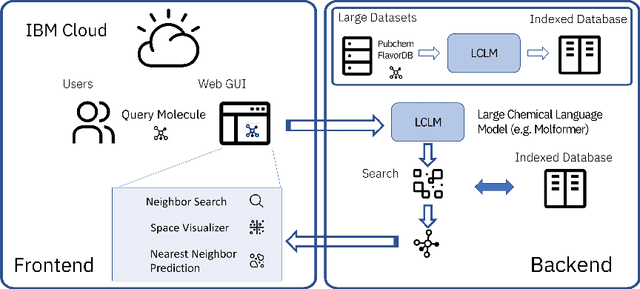
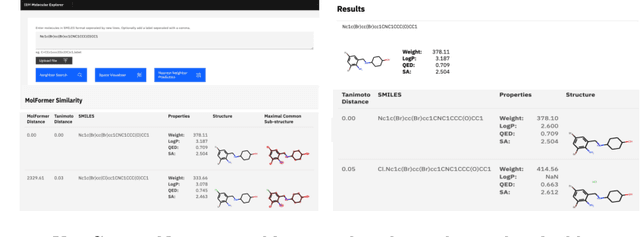
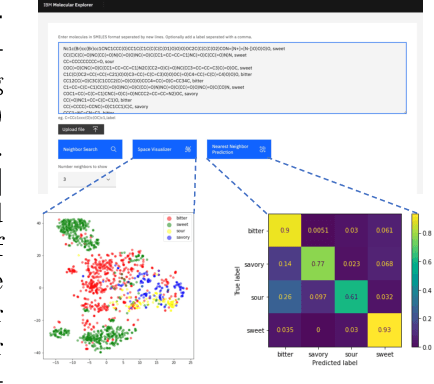
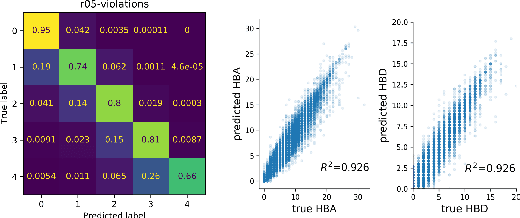
With the prospect of automating a number of chemical tasks with high fidelity, chemical language processing models are emerging at a rapid speed. Here, we present a cloud-based real-time platform that allows users to virtually screen molecules of interest. For this purpose, molecular embeddings inferred from a recently proposed large chemical language model, named MolFormer, are leveraged. The platform currently supports three tasks: nearest neighbor retrieval, chemical space visualization, and property prediction. Based on the functionalities of this platform and results obtained, we believe that such a platform can play a pivotal role in automating chemistry and chemical engineering research, as well as assist in drug discovery and material design tasks. A demo of our platform is provided at \url{www.ibm.biz/molecular_demo}.
GT4SD: Generative Toolkit for Scientific Discovery
Jul 08, 2022
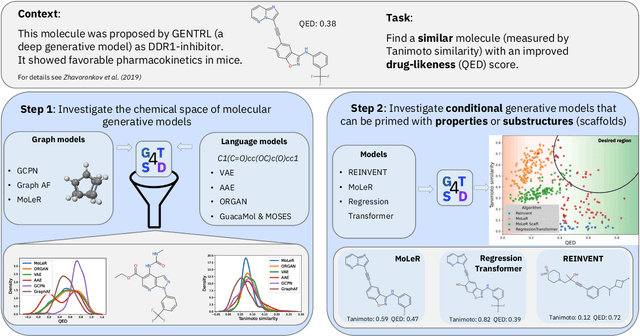
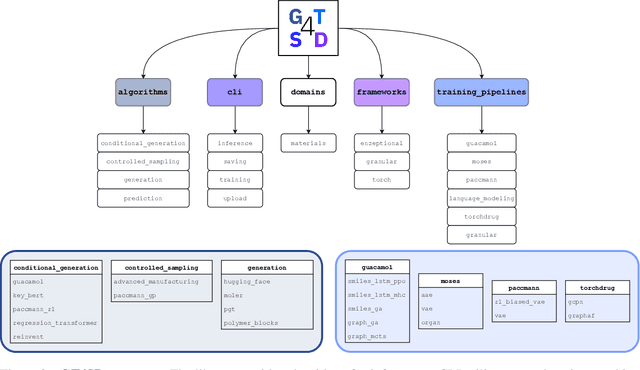
With the growing availability of data within various scientific domains, generative models hold enormous potential to accelerate scientific discovery at every step of the scientific method. Perhaps their most valuable application lies in the speeding up of what has traditionally been the slowest and most challenging step of coming up with a hypothesis. Powerful representations are now being learned from large volumes of data to generate novel hypotheses, which is making a big impact on scientific discovery applications ranging from material design to drug discovery. The GT4SD (https://github.com/GT4SD/gt4sd-core) is an extensible open-source library that enables scientists, developers and researchers to train and use state-of-the-art generative models for hypothesis generation in scientific discovery. GT4SD supports a variety of uses of generative models across material science and drug discovery, including molecule discovery and design based on properties related to target proteins, omic profiles, scaffold distances, binding energies and more.
Learning Geometrically Disentangled Representations of Protein Folding Simulations
May 20, 2022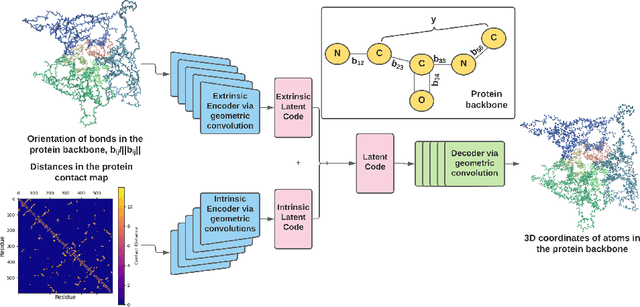
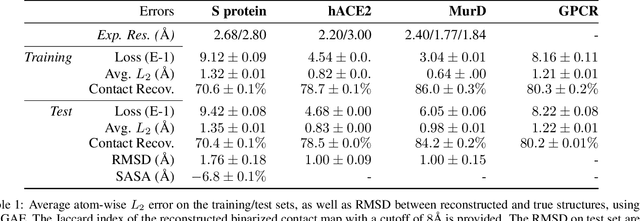
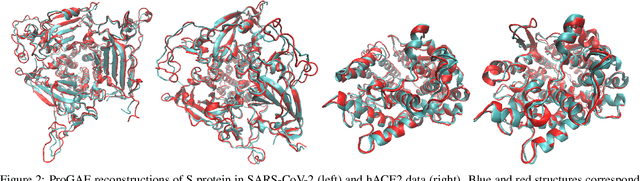

Massive molecular simulations of drug-target proteins have been used as a tool to understand disease mechanism and develop therapeutics. This work focuses on learning a generative neural network on a structural ensemble of a drug-target protein, e.g. SARS-CoV-2 Spike protein, obtained from computationally expensive molecular simulations. Model tasks involve characterizing the distinct structural fluctuations of the protein bound to various drug molecules, as well as efficient generation of protein conformations that can serve as an complement of a molecular simulation engine. Specifically, we present a geometric autoencoder framework to learn separate latent space encodings of the intrinsic and extrinsic geometries of the protein structure. For this purpose, the proposed Protein Geometric AutoEncoder (ProGAE) model is trained on the protein contact map and the orientation of the backbone bonds of the protein. Using ProGAE latent embeddings, we reconstruct and generate the conformational ensemble of a protein at or near the experimental resolution, while gaining better interpretability and controllability in term of protein structure generation from the learned latent space. Additionally, ProGAE models are transferable to a different state of the same protein or to a new protein of different size, where only the dense layer decoding from the latent representation needs to be retrained. Results show that our geometric learning-based method enjoys both accuracy and efficiency for generating complex structural variations, charting the path toward scalable and improved approaches for analyzing and enhancing high-cost simulations of drug-target proteins.
Accelerating Inhibitor Discovery for Multiple SARS-CoV-2 Targets with a Single, Sequence-Guided Deep Generative Framework
Apr 19, 2022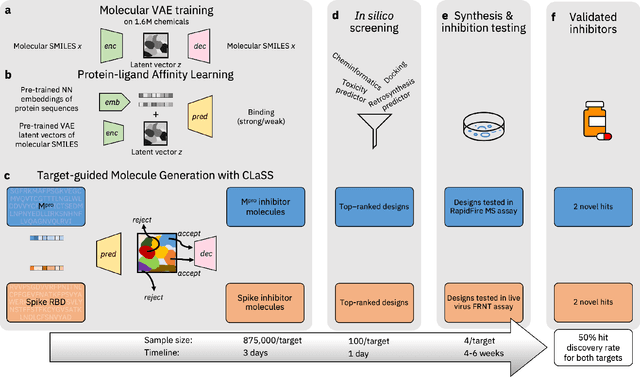


The COVID-19 pandemic has highlighted the urgency for developing more efficient molecular discovery pathways. As exhaustive exploration of the vast chemical space is infeasible, discovering novel inhibitor molecules for emerging drug-target proteins is challenging, particularly for targets with unknown structure or ligands. We demonstrate the broad utility of a single deep generative framework toward discovering novel drug-like inhibitor molecules against two distinct SARS-CoV-2 targets -- the main protease (Mpro) and the receptor binding domain (RBD) of the spike protein. To perform target-aware design, the framework employs a target sequence-conditioned sampling of novel molecules from a generative model. Micromolar-level in vitro inhibition was observed for two candidates (out of four synthesized) for each target. The most potent spike RBD inhibitor also emerged as a rare non-covalent antiviral with broad-spectrum activity against several SARS-CoV-2 variants in live virus neutralization assays. These results show a broadly deployable machine intelligence framework can accelerate hit discovery across different emerging drug-targets.
Protein Representation Learning by Geometric Structure Pretraining
Mar 14, 2022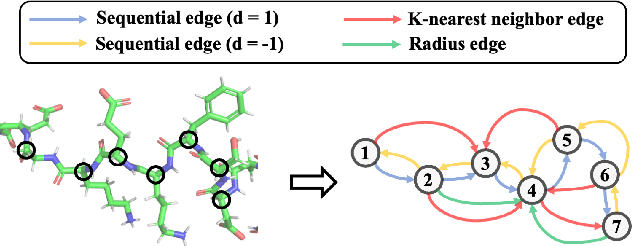
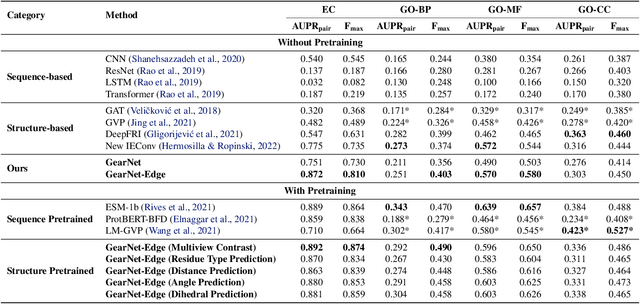
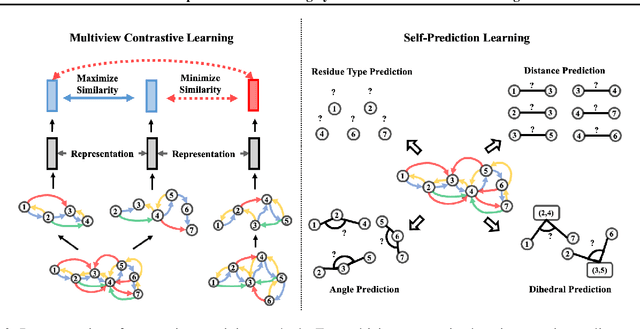
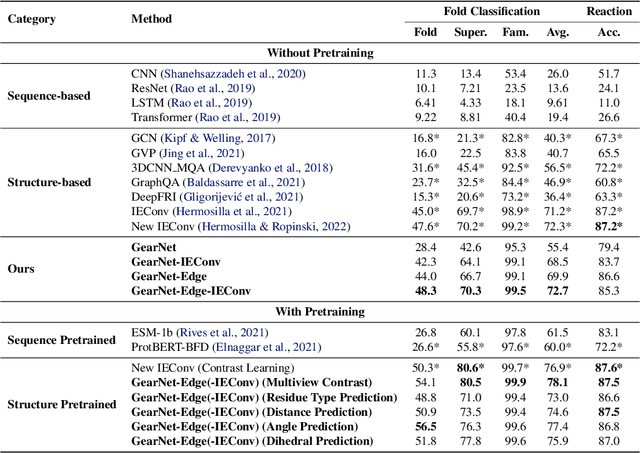
Learning effective protein representations is critical in a variety of tasks in biology such as predicting protein function or structure. Existing approaches usually pretrain protein language models on a large number of unlabeled amino acid sequences and then finetune the models with some labeled data in downstream tasks. Despite the effectiveness of sequence-based approaches, the power of pretraining on smaller numbers of known protein structures has not been explored for protein property prediction, though protein structures are known to be determinants of protein function. We first present a simple yet effective encoder to learn protein geometry features. We pretrain the protein graph encoder by leveraging multiview contrastive learning and different self-prediction tasks. Experimental results on both function prediction and fold classification tasks show that our proposed pretraining methods outperform or are on par with the state-of-the-art sequence-based methods using much less data. All codes and models will be published upon acceptance.
Sample-Efficient Generation of Novel Photo-acid Generator Molecules using a Deep Generative Model
Dec 02, 2021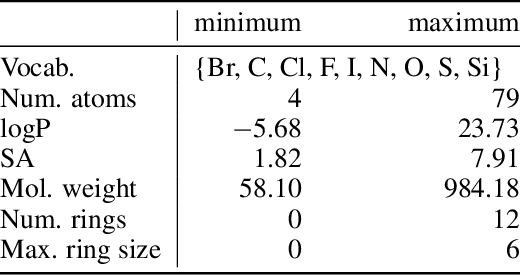
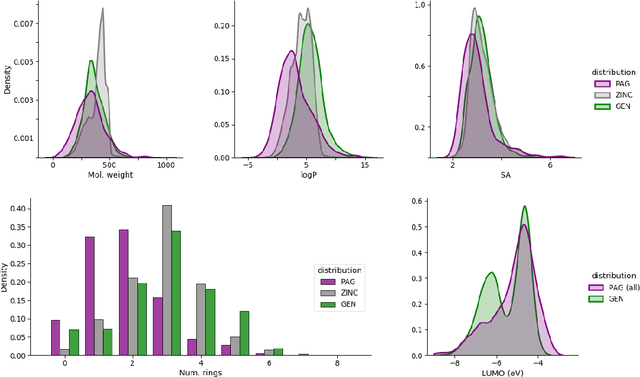
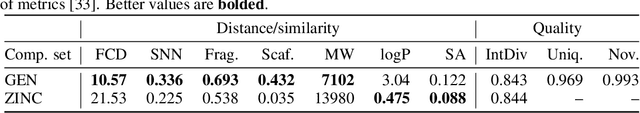
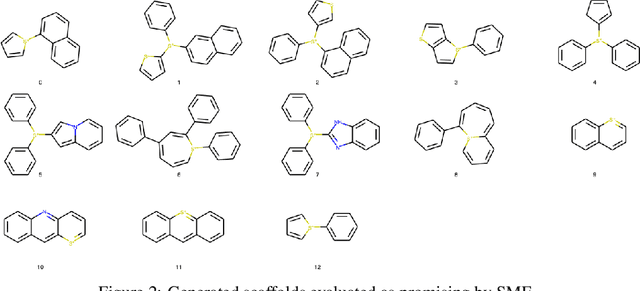
Photo-acid generators (PAGs) are compounds that release acids ($H^+$ ions) when exposed to light. These compounds are critical components of the photolithography processes that are used in the manufacture of semiconductor logic and memory chips. The exponential increase in the demand for semiconductors has highlighted the need for discovering novel photo-acid generators. While de novo molecule design using deep generative models has been widely employed for drug discovery and material design, its application to the creation of novel photo-acid generators poses several unique challenges, such as lack of property labels. In this paper, we highlight these challenges and propose a generative modeling approach that utilizes conditional generation from a pre-trained deep autoencoder and expert-in-the-loop techniques. The validity of the proposed approach was evaluated with the help of subject matter experts, indicating the promise of such an approach for applications beyond the creation of novel photo-acid generators.
Benchmarking deep generative models for diverse antibody sequence design
Nov 12, 2021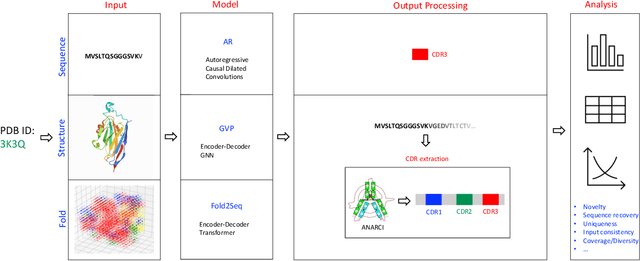

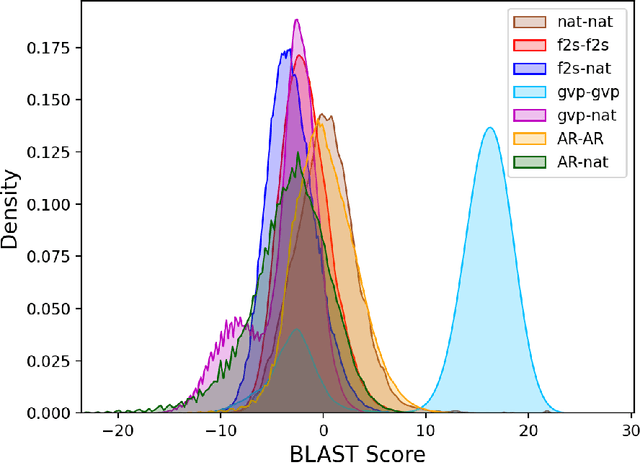
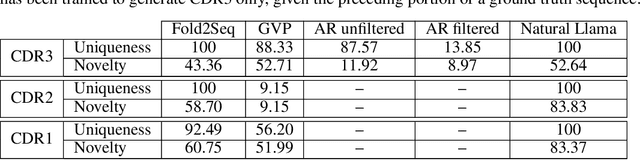
Computational protein design, i.e. inferring novel and diverse protein sequences consistent with a given structure, remains a major unsolved challenge. Recently, deep generative models that learn from sequences alone or from sequences and structures jointly have shown impressive performance on this task. However, those models appear limited in terms of modeling structural constraints, capturing enough sequence diversity, or both. Here we consider three recently proposed deep generative frameworks for protein design: (AR) the sequence-based autoregressive generative model, (GVP) the precise structure-based graph neural network, and Fold2Seq that leverages a fuzzy and scale-free representation of a three-dimensional fold, while enforcing structure-to-sequence (and vice versa) consistency. We benchmark these models on the task of computational design of antibody sequences, which demand designing sequences with high diversity for functional implication. The Fold2Seq framework outperforms the two other baselines in terms of diversity of the designed sequences, while maintaining the typical fold.
Fold2Seq: A Joint Sequence(1D)-Fold(3D) Embedding-based Generative Model for Protein Design
Jun 24, 2021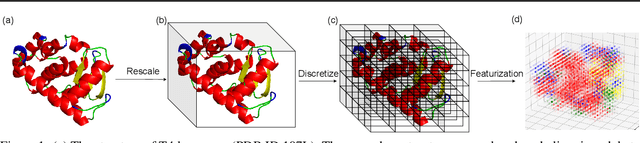
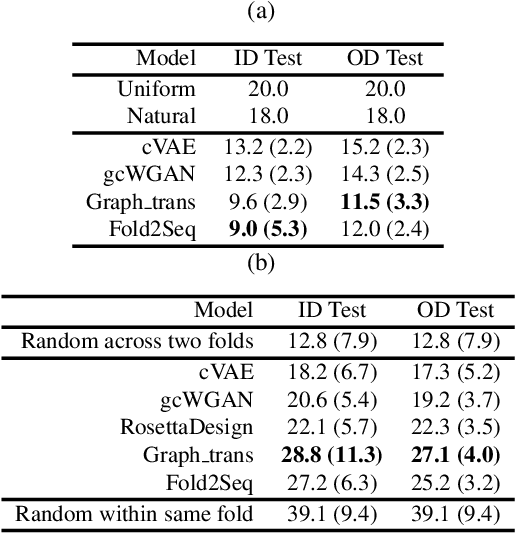
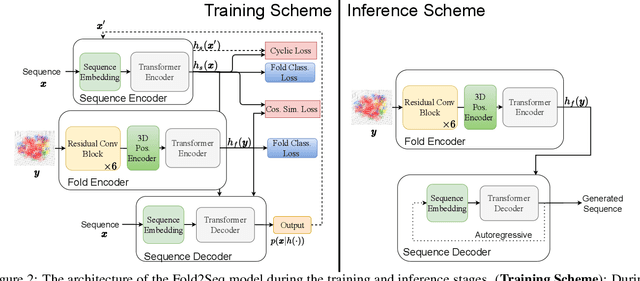
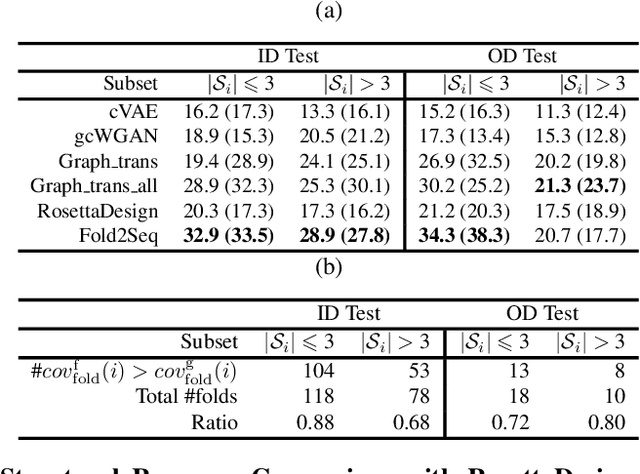
Designing novel protein sequences for a desired 3D topological fold is a fundamental yet non-trivial task in protein engineering. Challenges exist due to the complex sequence--fold relationship, as well as the difficulties to capture the diversity of the sequences (therefore structures and functions) within a fold. To overcome these challenges, we propose Fold2Seq, a novel transformer-based generative framework for designing protein sequences conditioned on a specific target fold. To model the complex sequence--structure relationship, Fold2Seq jointly learns a sequence embedding using a transformer and a fold embedding from the density of secondary structural elements in 3D voxels. On test sets with single, high-resolution and complete structure inputs for individual folds, our experiments demonstrate improved or comparable performance of Fold2Seq in terms of speed, coverage, and reliability for sequence design, when compared to existing state-of-the-art methods that include data-driven deep generative models and physics-based RosettaDesign. The unique advantages of fold-based Fold2Seq, in comparison to a structure-based deep model and RosettaDesign, become more evident on three additional real-world challenges originating from low-quality, incomplete, or ambiguous input structures. Source code and data are available at https://github.com/IBM/fold2seq.
Do Large Scale Molecular Language Representations Capture Important Structural Information?
Jun 17, 2021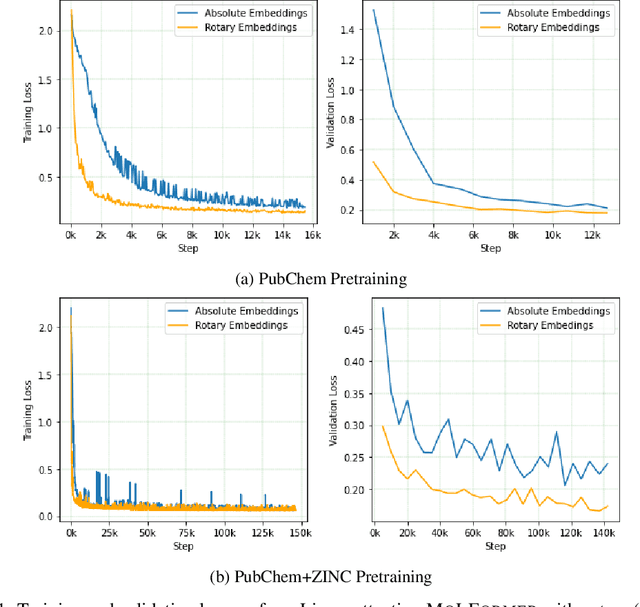
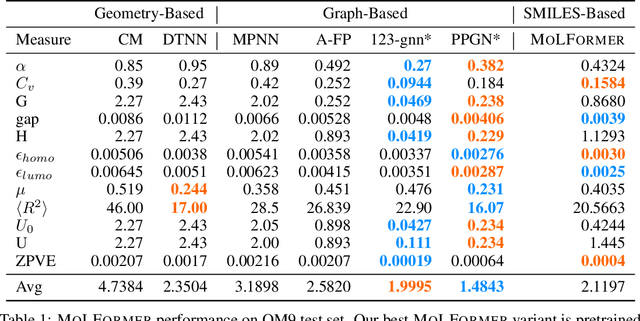
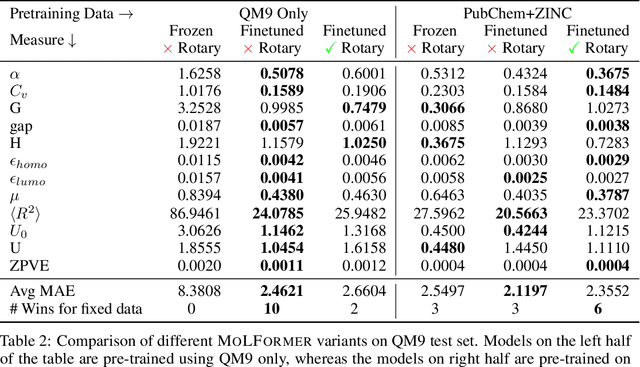
Predicting chemical properties from the structure of a molecule is of great importance in many applications including drug discovery and material design. Machine learning based molecular property prediction holds the promise of enabling accurate predictions at much less complexity, when compared to, for example Density Functional Theory (DFT) calculations. Features extracted from molecular graphs, using graph neural nets in a supervised manner, have emerged as strong baselines for such tasks. However, the vast chemical space together with the limited availability of labels makes supervised learning challenging, calling for learning a general-purpose molecular representation. Recently, pre-trained transformer-based language models (PTLMs) on large unlabeled corpus have produced state-of-the-art results in many downstream natural language processing tasks. Inspired by this development, here we present molecular embeddings obtained by training an efficient transformer encoder model, referred to as MoLFormer. This model was employed with a linear attention mechanism and highly paralleized training on 1D SMILES sequences of 1.1 billion unlabeled molecules from the PubChem and ZINC datasets. Experiments show that the learned molecular representation performs competitively, when compared to existing graph-based and fingerprint-based supervised learning baselines, on the challenging tasks of predicting properties of QM8 and QM9 molecules. Further task-specific fine-tuning of the MoLFormerr representation improves performance on several of those property prediction benchmarks. These results provide encouraging evidence that large-scale molecular language models can capture sufficient structural information to be able to accurately predict quantum chemical properties and beyond.