 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFunction Composition in Trustworthy Machine Learning: Implementation Choices, Insights, and Questions
Feb 17, 2023



Ensuring trustworthiness in machine learning (ML) models is a multi-dimensional task. In addition to the traditional notion of predictive performance, other notions such as privacy, fairness, robustness to distribution shift, adversarial robustness, interpretability, explainability, and uncertainty quantification are important considerations to evaluate and improve (if deficient). However, these sub-disciplines or 'pillars' of trustworthiness have largely developed independently, which has limited us from understanding their interactions in real-world ML pipelines. In this paper, focusing specifically on compositions of functions arising from the different pillars, we aim to reduce this gap, develop new insights for trustworthy ML, and answer questions such as the following. Does the composition of multiple fairness interventions result in a fairer model compared to a single intervention? How do bias mitigation algorithms for fairness affect local post-hoc explanations? Does a defense algorithm for untargeted adversarial attacks continue to be effective when composed with a privacy transformation? Toward this end, we report initial empirical results and new insights from 9 different compositions of functions (or pipelines) on 7 real-world datasets along two trustworthy dimensions - fairness and explainability. We also report progress, and implementation choices, on an extensible composer tool to encourage the combination of functionalities from multiple pillars. To-date, the tool supports bias mitigation algorithms for fairness and post-hoc explainability methods. We hope this line of work encourages the thoughtful consideration of multiple pillars when attempting to formulate and resolve a trustworthiness problem.
Navigating Ensemble Configurations for Algorithmic Fairness
Oct 11, 2022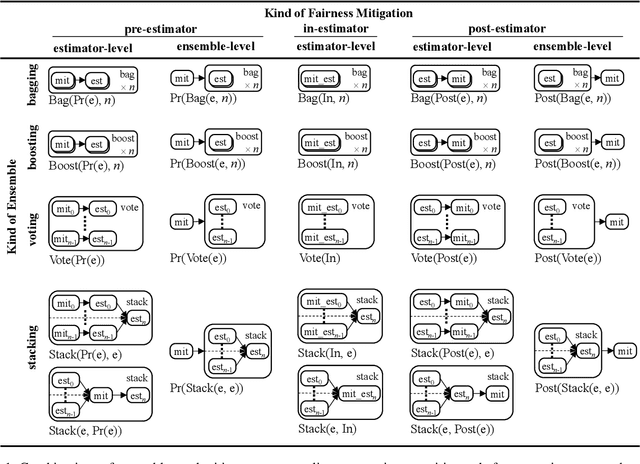

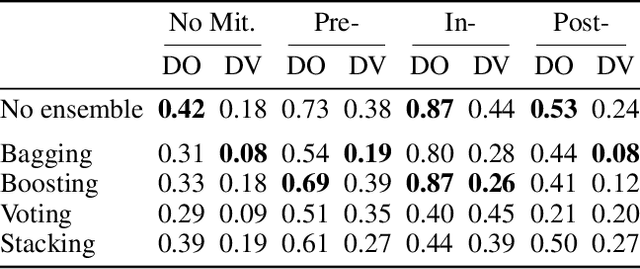
Bias mitigators can improve algorithmic fairness in machine learning models, but their effect on fairness is often not stable across data splits. A popular approach to train more stable models is ensemble learning, but unfortunately, it is unclear how to combine ensembles with mitigators to best navigate trade-offs between fairness and predictive performance. To that end, we built an open-source library enabling the modular composition of 8 mitigators, 4 ensembles, and their corresponding hyperparameters, and we empirically explored the space of configurations on 13 datasets. We distilled our insights from this exploration in the form of a guidance diagram for practitioners that we demonstrate is robust and reproducible.
Causal Graphs Underlying Generative Models: Path to Learning with Limited Data
Jul 14, 2022

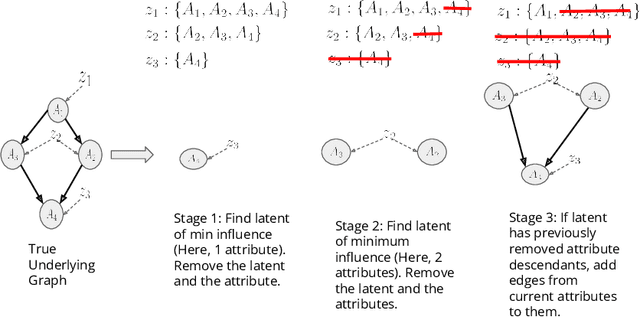

Training generative models that capture rich semantics of the data and interpreting the latent representations encoded by such models are very important problems in unsupervised learning. In this work, we provide a simple algorithm that relies on perturbation experiments on latent codes of a pre-trained generative autoencoder to uncover a causal graph that is implied by the generative model. We leverage pre-trained attribute classifiers and perform perturbation experiments to check for influence of a given latent variable on a subset of attributes. Given this, we show that one can fit an effective causal graph that models a structural equation model between latent codes taken as exogenous variables and attributes taken as observed variables. One interesting aspect is that a single latent variable controls multiple overlapping subsets of attributes unlike conventional approach that tries to impose full independence. Using a pre-trained RNN-based generative autoencoder trained on a dataset of peptide sequences, we demonstrate that the learnt causal graph from our algorithm between various attributes and latent codes can be used to predict a specific property for sequences which are unseen. We compare prediction models trained on either all available attributes or only the ones in the Markov blanket and empirically show that in both the unsupervised and supervised regimes, typically, using the predictor that relies on Markov blanket attributes generalizes better for out-of-distribution sequences.
GT4SD: Generative Toolkit for Scientific Discovery
Jul 08, 2022
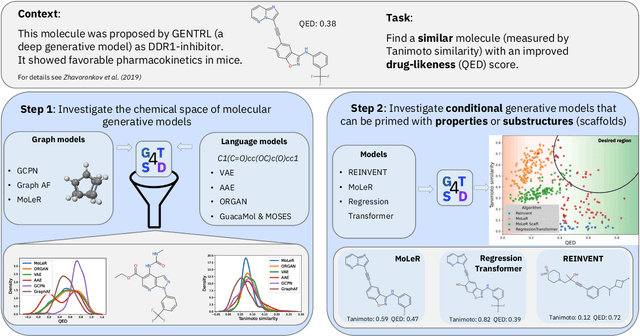
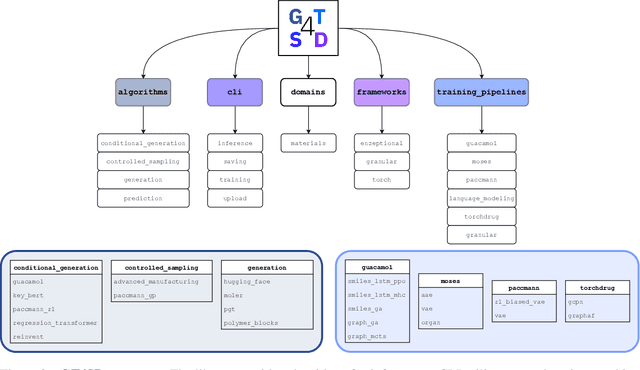
With the growing availability of data within various scientific domains, generative models hold enormous potential to accelerate scientific discovery at every step of the scientific method. Perhaps their most valuable application lies in the speeding up of what has traditionally been the slowest and most challenging step of coming up with a hypothesis. Powerful representations are now being learned from large volumes of data to generate novel hypotheses, which is making a big impact on scientific discovery applications ranging from material design to drug discovery. The GT4SD (https://github.com/GT4SD/gt4sd-core) is an extensible open-source library that enables scientists, developers and researchers to train and use state-of-the-art generative models for hypothesis generation in scientific discovery. GT4SD supports a variety of uses of generative models across material science and drug discovery, including molecule discovery and design based on properties related to target proteins, omic profiles, scaffold distances, binding energies and more.
Accelerating Inhibitor Discovery for Multiple SARS-CoV-2 Targets with a Single, Sequence-Guided Deep Generative Framework
Apr 19, 2022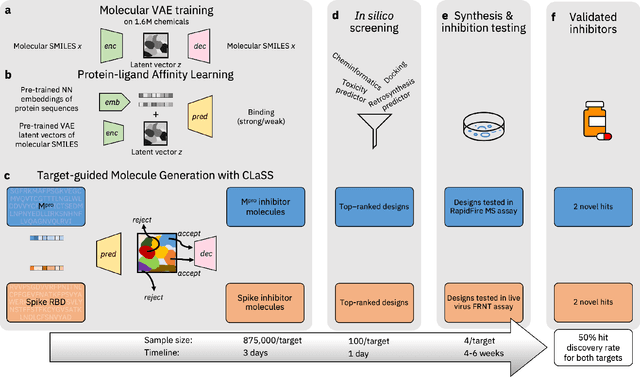

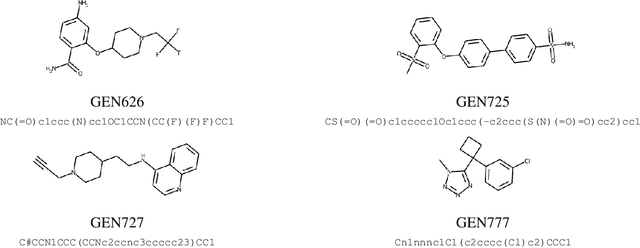

The COVID-19 pandemic has highlighted the urgency for developing more efficient molecular discovery pathways. As exhaustive exploration of the vast chemical space is infeasible, discovering novel inhibitor molecules for emerging drug-target proteins is challenging, particularly for targets with unknown structure or ligands. We demonstrate the broad utility of a single deep generative framework toward discovering novel drug-like inhibitor molecules against two distinct SARS-CoV-2 targets -- the main protease (Mpro) and the receptor binding domain (RBD) of the spike protein. To perform target-aware design, the framework employs a target sequence-conditioned sampling of novel molecules from a generative model. Micromolar-level in vitro inhibition was observed for two candidates (out of four synthesized) for each target. The most potent spike RBD inhibitor also emerged as a rare non-covalent antiviral with broad-spectrum activity against several SARS-CoV-2 variants in live virus neutralization assays. These results show a broadly deployable machine intelligence framework can accelerate hit discovery across different emerging drug-targets.
An Empirical Study of Modular Bias Mitigators and Ensembles
Feb 01, 2022
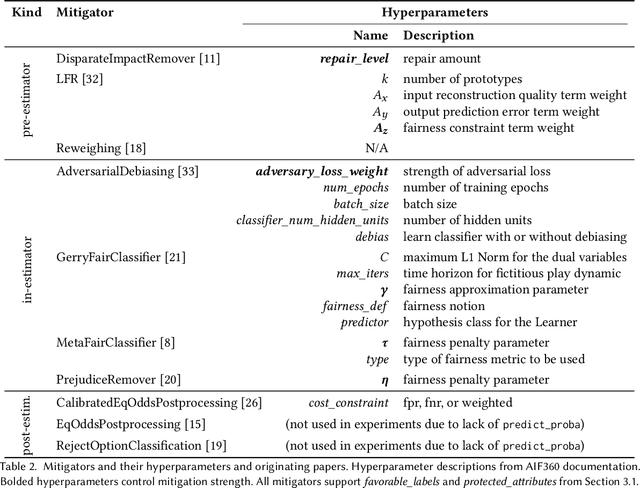

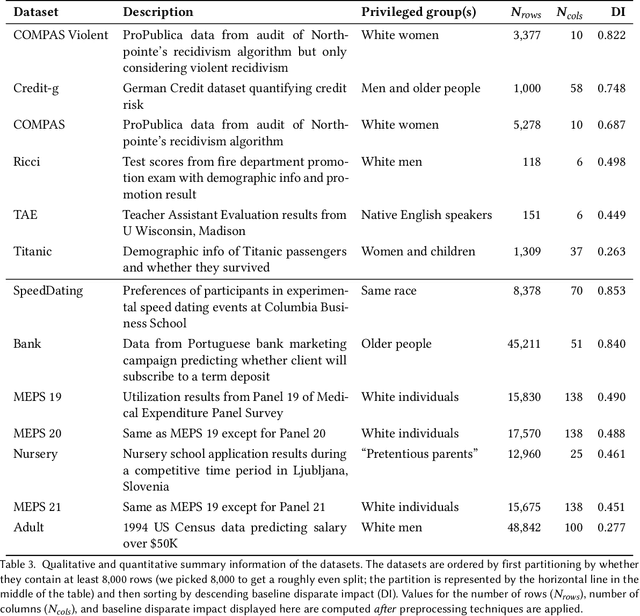
There are several bias mitigators that can reduce algorithmic bias in machine learning models but, unfortunately, the effect of mitigators on fairness is often not stable when measured across different data splits. A popular approach to train more stable models is ensemble learning. Ensembles, such as bagging, boosting, voting, or stacking, have been successful at making predictive performance more stable. One might therefore ask whether we can combine the advantages of bias mitigators and ensembles? To explore this question, we first need bias mitigators and ensembles to work together. We built an open-source library enabling the modular composition of 10 mitigators, 4 ensembles, and their corresponding hyperparameters. Based on this library, we empirically explored the space of combinations on 13 datasets, including datasets commonly used in fairness literature plus datasets newly curated by our library. Furthermore, we distilled the results into a guidance diagram for practitioners. We hope this paper will contribute towards improving stability in bias mitigation.
Sample-Efficient Generation of Novel Photo-acid Generator Molecules using a Deep Generative Model
Dec 02, 2021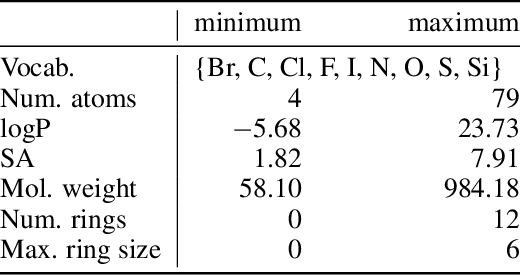
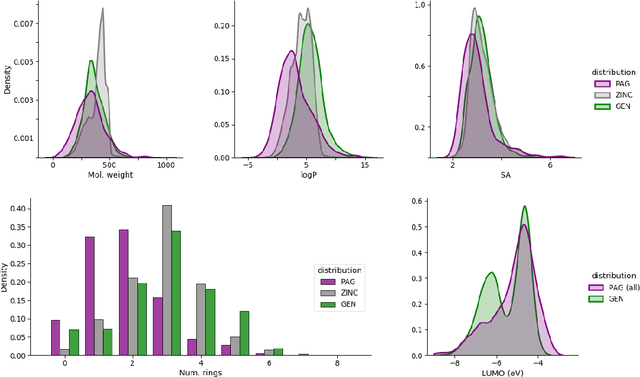
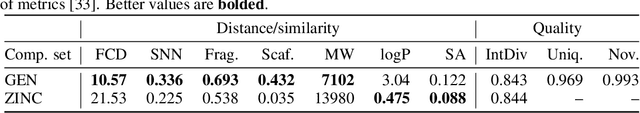
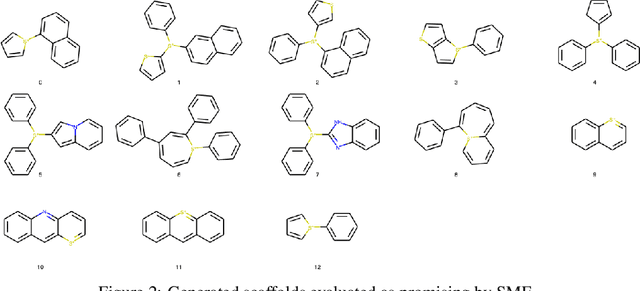
Photo-acid generators (PAGs) are compounds that release acids ($H^+$ ions) when exposed to light. These compounds are critical components of the photolithography processes that are used in the manufacture of semiconductor logic and memory chips. The exponential increase in the demand for semiconductors has highlighted the need for discovering novel photo-acid generators. While de novo molecule design using deep generative models has been widely employed for drug discovery and material design, its application to the creation of novel photo-acid generators poses several unique challenges, such as lack of property labels. In this paper, we highlight these challenges and propose a generative modeling approach that utilizes conditional generation from a pre-trained deep autoencoder and expert-in-the-loop techniques. The validity of the proposed approach was evaluated with the help of subject matter experts, indicating the promise of such an approach for applications beyond the creation of novel photo-acid generators.
AI Explainability 360: Impact and Design
Sep 24, 2021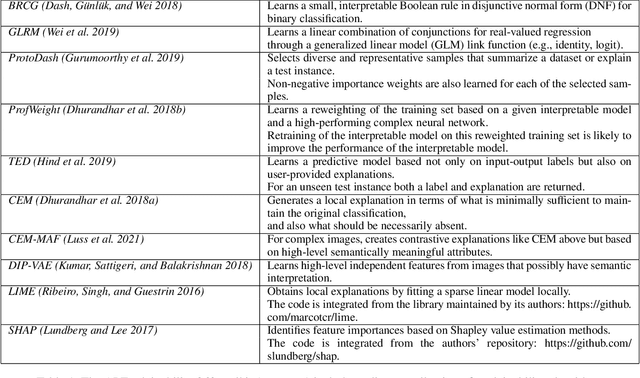
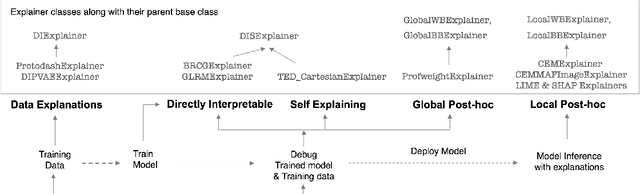

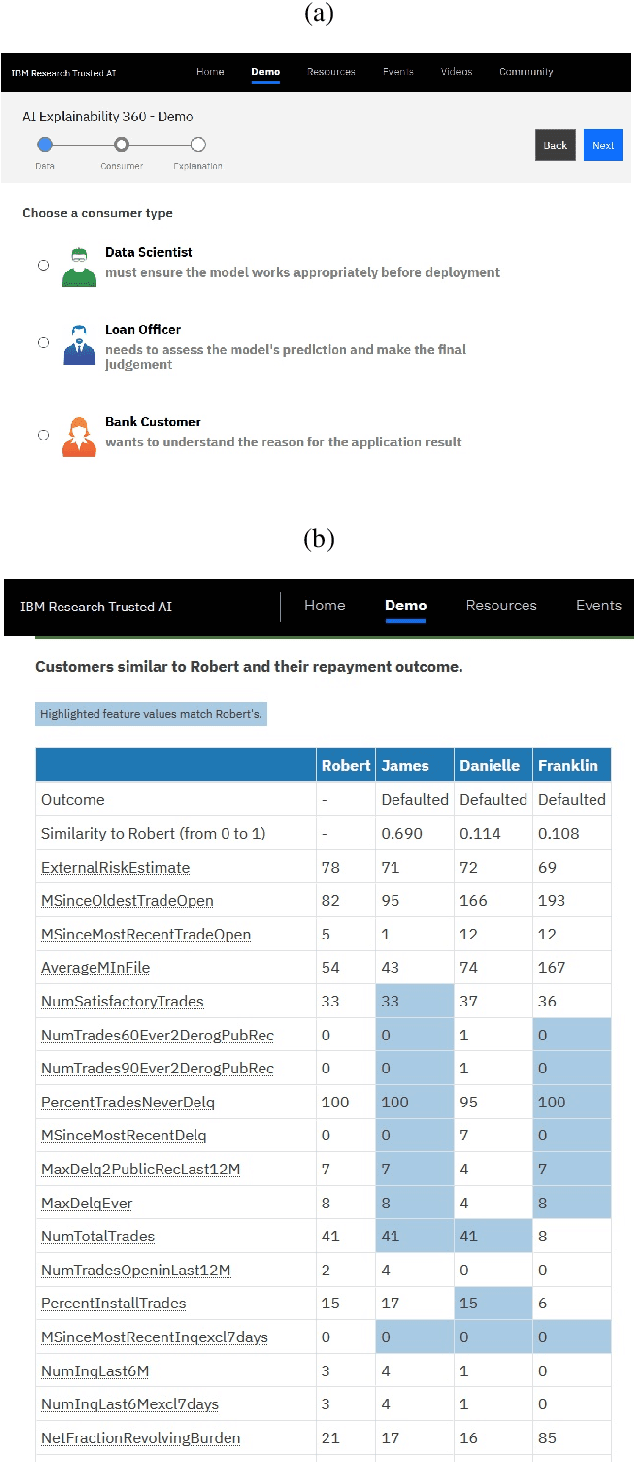
As artificial intelligence and machine learning algorithms become increasingly prevalent in society, multiple stakeholders are calling for these algorithms to provide explanations. At the same time, these stakeholders, whether they be affected citizens, government regulators, domain experts, or system developers, have different explanation needs. To address these needs, in 2019, we created AI Explainability 360 (Arya et al. 2020), an open source software toolkit featuring ten diverse and state-of-the-art explainability methods and two evaluation metrics. This paper examines the impact of the toolkit with several case studies, statistics, and community feedback. The different ways in which users have experienced AI Explainability 360 have resulted in multiple types of impact and improvements in multiple metrics, highlighted by the adoption of the toolkit by the independent LF AI & Data Foundation. The paper also describes the flexible design of the toolkit, examples of its use, and the significant educational material and documentation available to its users.
Target-Specific and Selective Drug Design for COVID-19 Using Deep Generative Models
Apr 02, 2020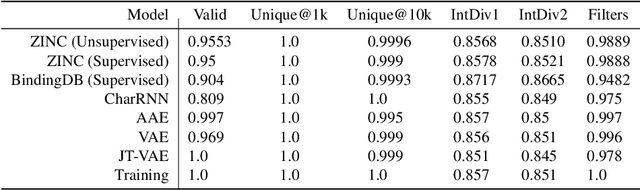
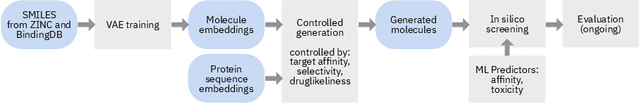
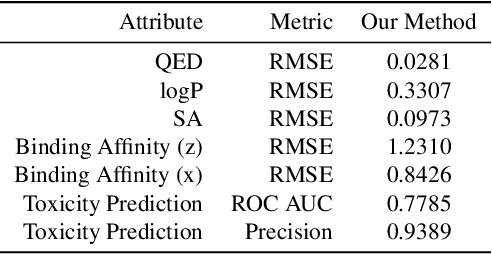
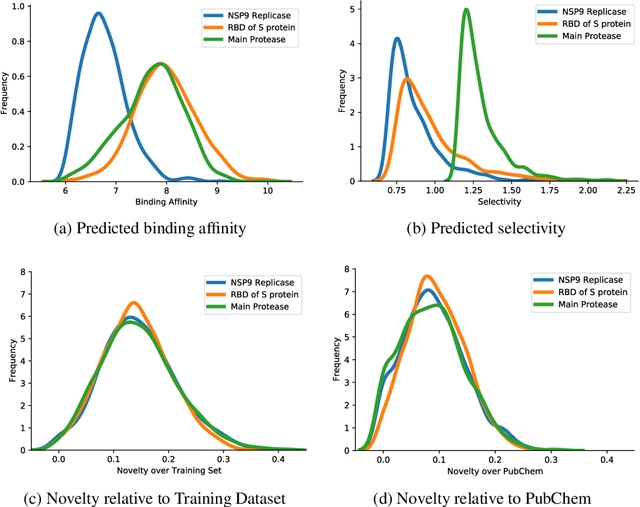
The recent COVID-19 pandemic has highlighted the need for rapid therapeutic development for infectious diseases. To accelerate this process, we present a deep learning based generative modeling framework, CogMol, to design drug candidates specific to a given target protein sequence with high off-target selectivity. We augment this generative framework with an in silico screening process that accounts for toxicity, to lower the failure rate of the generated drug candidates in later stages of the drug development pipeline. We apply this framework to three relevant proteins of the SARS-CoV-2, the virus responsible for COVID-19, namely non-structural protein 9 (NSP9) replicase, main protease, and the receptor-binding domain (RBD) of the S protein. Docking to the target proteins demonstrate the potential of these generated molecules as ligands. Structural similarity analyses further imply novelty of the generated molecules with respect to the training dataset as well as possible biological association of a number of generated molecules that might be of relevance to COVID-19 therapeutic design. While the validation of these molecules is underway, we release ~ 3000 novel COVID-19 drug candidates generated using our framework. URL : http://ibm.biz/covid19-mol
One Explanation Does Not Fit All: A Toolkit and Taxonomy of AI Explainability Techniques
Sep 14, 2019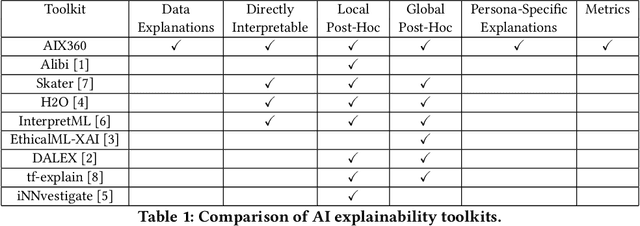
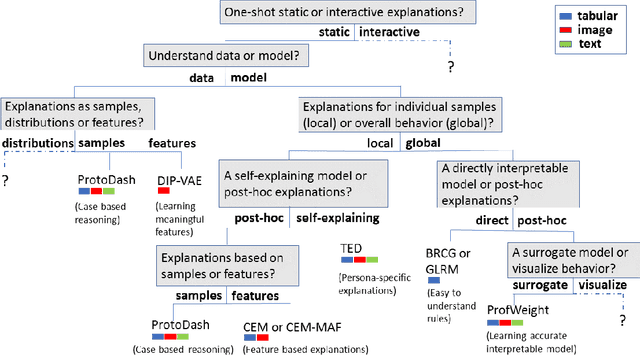
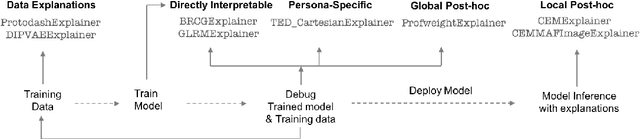
As artificial intelligence and machine learning algorithms make further inroads into society, calls are increasing from multiple stakeholders for these algorithms to explain their outputs. At the same time, these stakeholders, whether they be affected citizens, government regulators, domain experts, or system developers, present different requirements for explanations. Toward addressing these needs, we introduce AI Explainability 360 (http://aix360.mybluemix.net/), an open-source software toolkit featuring eight diverse and state-of-the-art explainability methods and two evaluation metrics. Equally important, we provide a taxonomy to help entities requiring explanations to navigate the space of explanation methods, not only those in the toolkit but also in the broader literature on explainability. For data scientists and other users of the toolkit, we have implemented an extensible software architecture that organizes methods according to their place in the AI modeling pipeline. We also discuss enhancements to bring research innovations closer to consumers of explanations, ranging from simplified, more accessible versions of algorithms, to tutorials and an interactive web demo to introduce AI explainability to different audiences and application domains. Together, our toolkit and taxonomy can help identify gaps where more explainability methods are needed and provide a platform to incorporate them as they are developed.