 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAdversarial Attack Framework on Graph Embedding Models with Limited Knowledge
May 26, 2021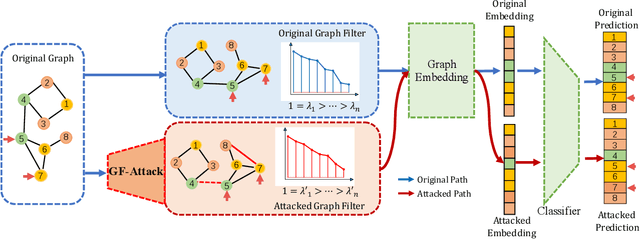

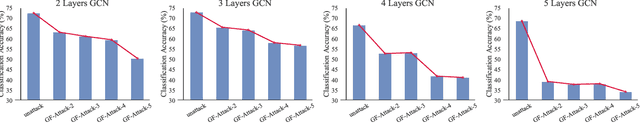
With the success of the graph embedding model in both academic and industry areas, the robustness of graph embedding against adversarial attack inevitably becomes a crucial problem in graph learning. Existing works usually perform the attack in a white-box fashion: they need to access the predictions/labels to construct their adversarial loss. However, the inaccessibility of predictions/labels makes the white-box attack impractical to a real graph learning system. This paper promotes current frameworks in a more general and flexible sense -- we demand to attack various kinds of graph embedding models with black-box driven. We investigate the theoretical connections between graph signal processing and graph embedding models and formulate the graph embedding model as a general graph signal process with a corresponding graph filter. Therefore, we design a generalized adversarial attacker: GF-Attack. Without accessing any labels and model predictions, GF-Attack can perform the attack directly on the graph filter in a black-box fashion. We further prove that GF-Attack can perform an effective attack without knowing the number of layers of graph embedding models. To validate the generalization of GF-Attack, we construct the attacker on four popular graph embedding models. Extensive experiments validate the effectiveness of GF-Attack on several benchmark datasets.
Recognizing Predictive Substructures with Subgraph Information Bottleneck
Mar 20, 2021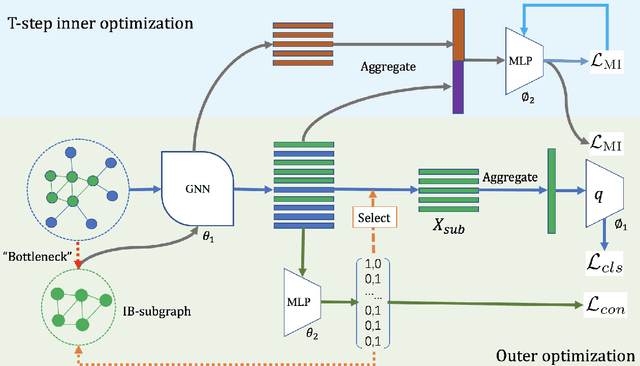
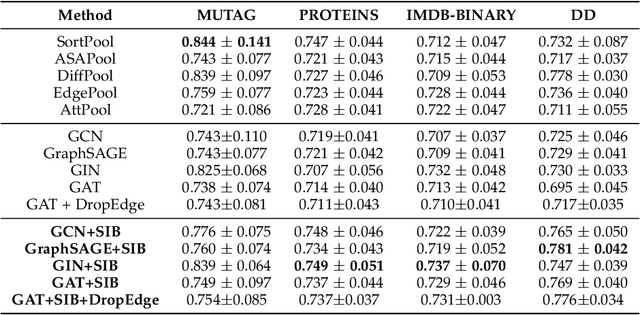

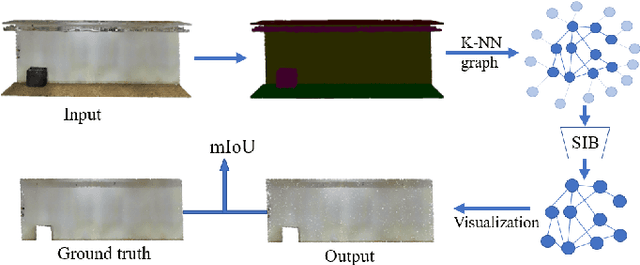
The emergence of Graph Convolutional Network (GCN) has greatly boosted the progress of graph learning. However, two disturbing factors, noise and redundancy in graph data, and lack of interpretation for prediction results, impede further development of GCN. One solution is to recognize a predictive yet compressed subgraph to get rid of the noise and redundancy and obtain the interpretable part of the graph. This setting of subgraph is similar to the information bottleneck (IB) principle, which is less studied on graph-structured data and GCN. Inspired by the IB principle, we propose a novel subgraph information bottleneck (SIB) framework to recognize such subgraphs, named IB-subgraph. However, the intractability of mutual information and the discrete nature of graph data makes the objective of SIB notoriously hard to optimize. To this end, we introduce a bilevel optimization scheme coupled with a mutual information estimator for irregular graphs. Moreover, we propose a continuous relaxation for subgraph selection with a connectivity loss for stabilization. We further theoretically prove the error bound of our estimation scheme for mutual information and the noise-invariant nature of IB-subgraph. Extensive experiments on graph learning and large-scale point cloud tasks demonstrate the superior property of IB-subgraph.
Diversified Multiscale Graph Learning with Graph Self-Correction
Mar 17, 2021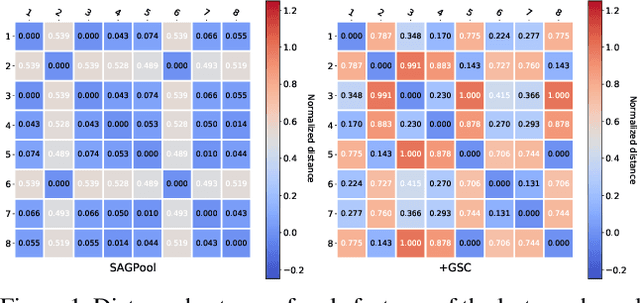
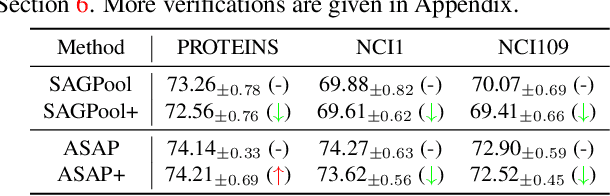
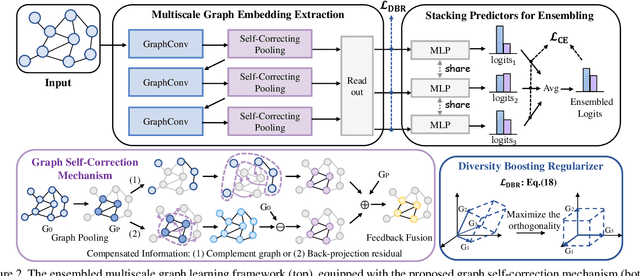
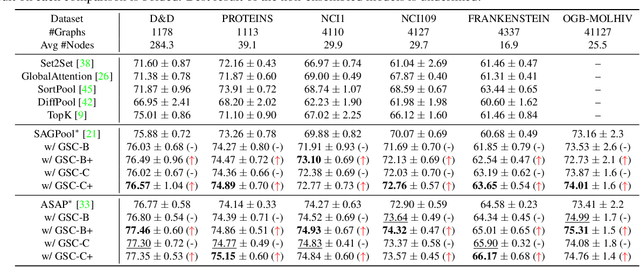
Though the multiscale graph learning techniques have enabled advanced feature extraction frameworks, the classic ensemble strategy may show inferior performance while encountering the high homogeneity of the learnt representation, which is caused by the nature of existing graph pooling methods. To cope with this issue, we propose a diversified multiscale graph learning model equipped with two core ingredients: a graph self-correction (GSC) mechanism to generate informative embedded graphs, and a diversity boosting regularizer (DBR) to achieve a comprehensive characterization of the input graph. The proposed GSC mechanism compensates the pooled graph with the lost information during the graph pooling process by feeding back the estimated residual graph, which serves as a plug-in component for popular graph pooling methods. Meanwhile, pooling methods enhanced with the GSC procedure encourage the discrepancy of node embeddings, and thus it contributes to the success of ensemble learning strategy. The proposed DBR instead enhances the ensemble diversity at the graph-level embeddings by leveraging the interaction among individual classifiers. Extensive experiments on popular graph classification benchmarks show that the proposed GSC mechanism leads to significant improvements over state-of-the-art graph pooling methods. Moreover, the ensemble multiscale graph learning models achieve superior enhancement by combining both GSC and DBR.
Deep Multimodal Fusion by Channel Exchanging
Nov 10, 2020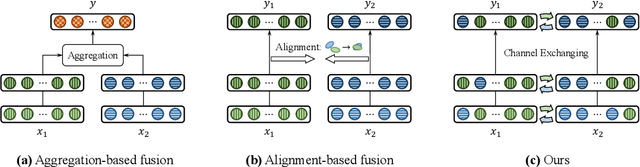
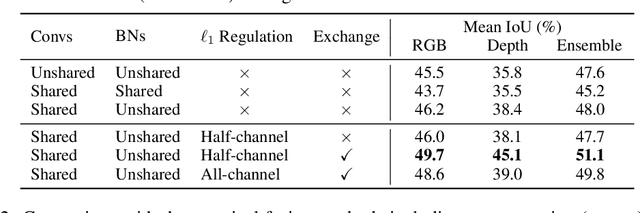
Deep multimodal fusion by using multiple sources of data for classification or regression has exhibited a clear advantage over the unimodal counterpart on various applications. Yet, current methods including aggregation-based and alignment-based fusion are still inadequate in balancing the trade-off between inter-modal fusion and intra-modal processing, incurring a bottleneck of performance improvement. To this end, this paper proposes Channel-Exchanging-Network (CEN), a parameter-free multimodal fusion framework that dynamically exchanges channels between sub-networks of different modalities. Specifically, the channel exchanging process is self-guided by individual channel importance that is measured by the magnitude of Batch-Normalization (BN) scaling factor during training. The validity of such exchanging process is also guaranteed by sharing convolutional filters yet keeping separate BN layers across modalities, which, as an add-on benefit, allows our multimodal architecture to be almost as compact as a unimodal network. Extensive experiments on semantic segmentation via RGB-D data and image translation through multi-domain input verify the effectiveness of our CEN compared to current state-of-the-art methods. Detailed ablation studies have also been carried out, which provably affirm the advantage of each component we propose. Our code is available at https://github.com/yikaiw/CEN.
On Self-Distilling Graph Neural Network
Nov 04, 2020
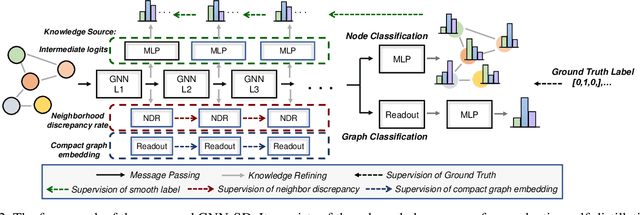
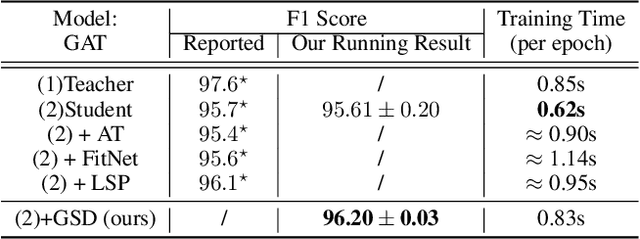
Recently, the teacher-student knowledge distillation framework has demonstrated its potential in training Graph Neural Networks (GNNs). However, due to the difficulty of training deep and wide GNN models, one can not always obtain a satisfactory teacher model for distillation. Furthermore, the inefficient training process of teacher-student knowledge distillation also impedes its applications in GNN models. In this paper, we propose the first teacher-free knowledge distillation framework for GNNs, termed GNN Self-Distillation (GNN-SD), that serves as a drop-in replacement for improving the training process of GNNs.We design three knowledge sources for GNN-SD: neighborhood discrepancy rate (NDR), compact graph embedding and intermediate logits. Notably, serving as a metric of the non-smoothness of the embedded graph, NDR empowers the transferability of knowledge that maintains high neighborhood discrepancy by enforcing consistency between consecutive GNN layers. We conduct exploring analysis to verify that our framework could improve the training dynamics and embedding quality of GNNs. Extensive experiments on various popular GNN models and datasets demonstrate that our approach obtains consistent and considerable performance enhancement, proving its effectiveness and generalization ability.
Graph Information Bottleneck for Subgraph Recognition
Oct 12, 2020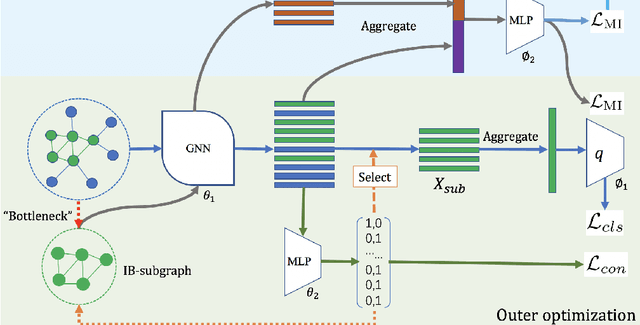
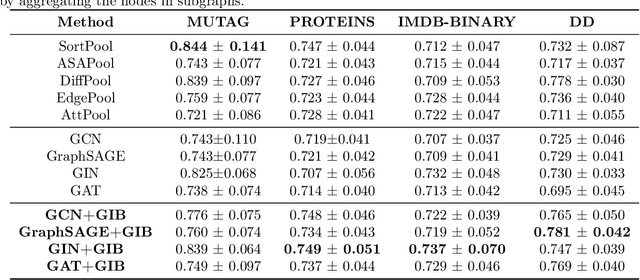
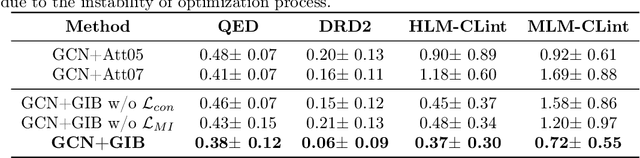
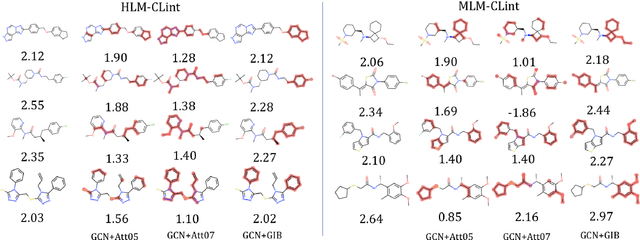
Given the input graph and its label/property, several key problems of graph learning, such as finding interpretable subgraphs, graph denoising and graph compression, can be attributed to the fundamental problem of recognizing a subgraph of the original one. This subgraph shall be as informative as possible, yet contains less redundant and noisy structure. This problem setting is closely related to the well-known information bottleneck (IB) principle, which, however, has less been studied for the irregular graph data and graph neural networks (GNNs). In this paper, we propose a framework of Graph Information Bottleneck (GIB) for the subgraph recognition problem in deep graph learning. Under this framework, one can recognize the maximally informative yet compressive subgraph, named IB-subgraph. However, the GIB objective is notoriously hard to optimize, mostly due to the intractability of the mutual information of irregular graph data and the unstable optimization process. In order to tackle these challenges, we propose: i) a GIB objective based-on a mutual information estimator for the irregular graph data; ii) a bi-level optimization scheme to maximize the GIB objective; iii) a connectivity loss to stabilize the optimization process. We evaluate the properties of the IB-subgraph in three application scenarios: improvement of graph classification, graph interpretation and graph denoising. Extensive experiments demonstrate that the information-theoretic IB-subgraph enjoys superior graph properties.
Tackling Over-Smoothing for General Graph Convolutional Networks
Sep 08, 2020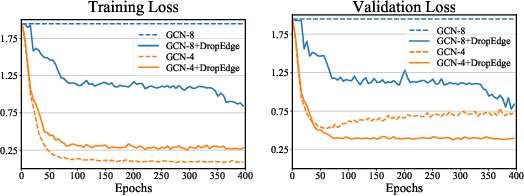
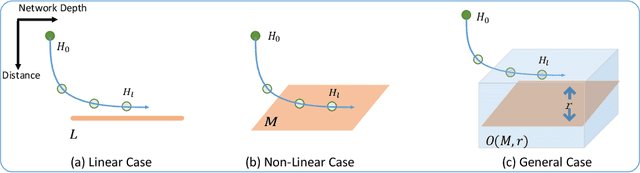
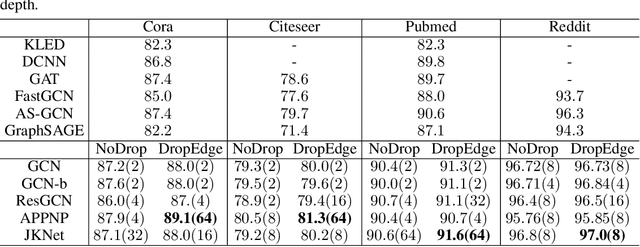
Increasing the depth of GCN, which is expected to permit more expressivity, is shown to incur performance detriment especially on node classification. The main cause of this lies in over-smoothing. The over-smoothing issue drives the output of GCN towards a space that contains limited distinguished information among nodes, leading to poor expressivity. Several works on refining the architecture of deep GCN have been proposed, but it is still unknown in theory whether or not these refinements are able to relieve over-smoothing. In this paper, we first theoretically analyze how general GCNs act with the increase in depth, including generic GCN, GCN with bias, ResGCN, and APPNP. We find that all these models are characterized by a universal process: all nodes converging to a cuboid. Upon this theorem, we propose DropEdge to alleviate over-smoothing by randomly removing a certain number of edges at each training epoch. Theoretically, DropEdge either reduces the convergence speed of over-smoothing or relieves the information loss caused by dimension collapse. Experimental evaluations on simulated dataset have visualized the difference in over-smoothing between different GCNs. Moreover, extensive experiments on several real benchmarks support that DropEdge consistently improves the performance on a variety of both shallow and deep GCNs.
Inverse Graph Identification: Can We Identify Node Labels Given Graph Labels?
Jul 12, 2020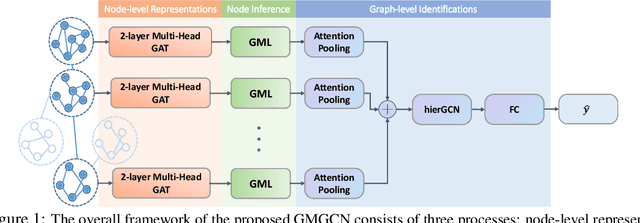
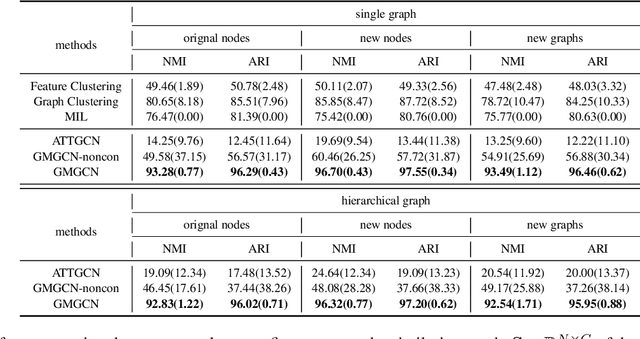
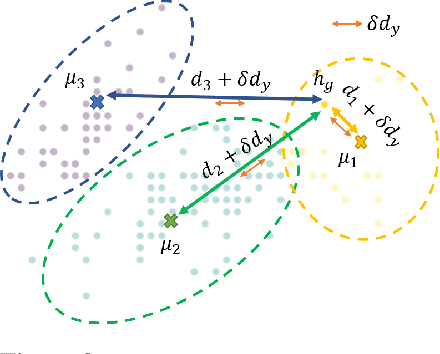
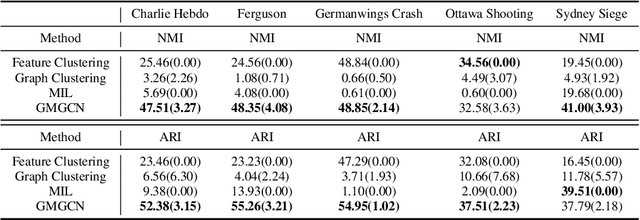
Graph Identification (GI) has long been researched in graph learning and is essential in certain applications (e.g. social community detection). Specifically, GI requires to predict the label/score of a target graph given its collection of node features and edge connections. While this task is common, more complex cases arise in practice---we are supposed to do the inverse thing by, for example, grouping similar users in a social network given the labels of different communities. This triggers an interesting thought: can we identify nodes given the labels of the graphs they belong to? Therefore, this paper defines a novel problem dubbed Inverse Graph Identification (IGI), as opposed to GI. Upon a formal discussion of the variants of IGI, we choose a particular case study of node clustering by making use of the graph labels and node features, with an assistance of a hierarchical graph that further characterizes the connections between different graphs. To address this task, we propose Gaussian Mixture Graph Convolutional Network (GMGCN), a simple yet effective method that makes the node-level message passing process using Graph Attention Network (GAT) under the protocol of GI and then infers the category of each node via a Gaussian Mixture Layer (GML). The training of GMGCN is further boosted by a proposed consensus loss to take advantage of the structure of the hierarchical graph. Extensive experiments are conducted to test the rationality of the formulation of IGI. We verify the superiority of the proposed method compared to other baselines on several benchmarks we have built up. We will release our codes along with the benchmark data to facilitate more research attention to the IGI problem.
GROVER: Self-supervised Message Passing Transformer on Large-scale Molecular Data
Jun 18, 2020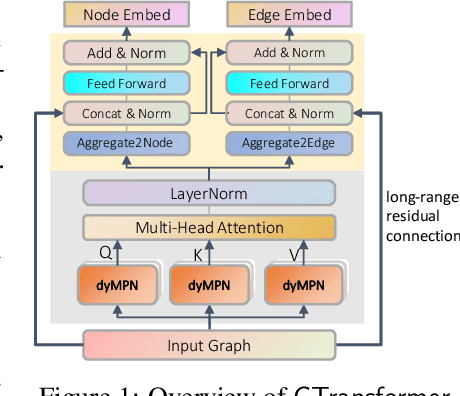
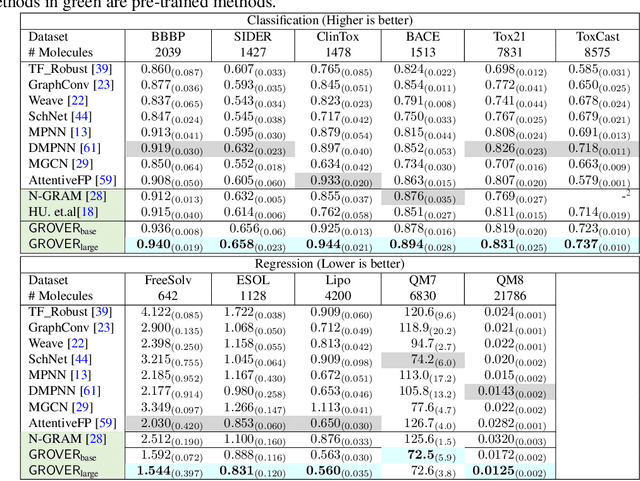
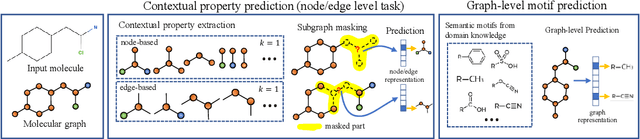
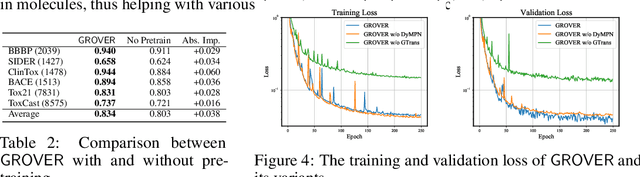
How to obtain informative representations of molecules is a crucial prerequisite in AI-driven drug design and discovery. Recent researches abstract molecules as graphs and employ Graph Neural Networks (GNNs) for task-specific and data-driven molecular representation learning. Nevertheless, two "dark clouds" impede the usage of GNNs in real scenarios: (1) insufficient labeled molecules for supervised training; (2) poor generalization capabilities to new-synthesized molecules. To address them both, we propose a novel molecular representation framework, GROVER, which stands for Graph Representation frOm self-superVised mEssage passing tRansformer. With carefully designed self-supervised tasks in node, edge and graph-level, GROVER can learn rich structural and semantic information of molecules from enormous unlabelled molecular data. Rather, to encode such complex information, GROVER integrates Message Passing Networks with the Transformer-style architecture to deliver a class of more expressive encoders of molecules. The flexibility of GROVER allows it to be trained efficiently on large-scale molecular dataset without requiring any supervision, thus being immunized to the two issues mentioned above. We pre-train GROVER with 100 million parameters on 10 million unlabelled molecules---the biggest GNN and the largest training dataset that we have ever met. We then leverage the pre-trained GROVER to downstream molecular property prediction tasks followed by task-specific fine-tuning, where we observe a huge improvement (more than 6% on average) over current state-of-the-art methods on 11 challenging benchmarks. The insights we gained are that well-designed self-supervision losses and largely-expressive pre-trained models enjoy the significant potential on performance boosting.
Multi-View Graph Neural Networks for Molecular Property Prediction
Jun 12, 2020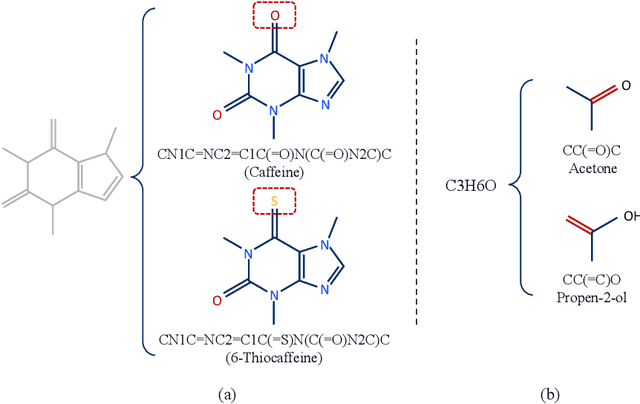
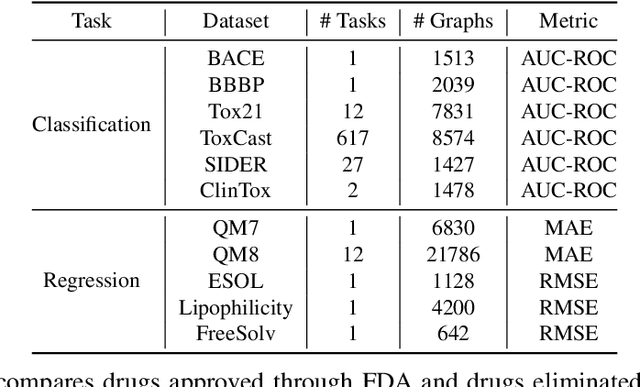
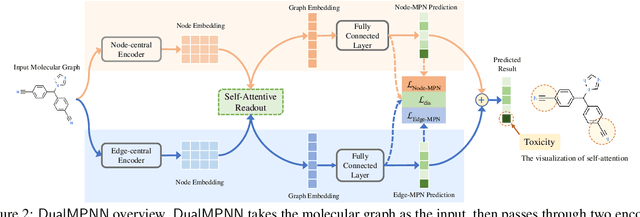
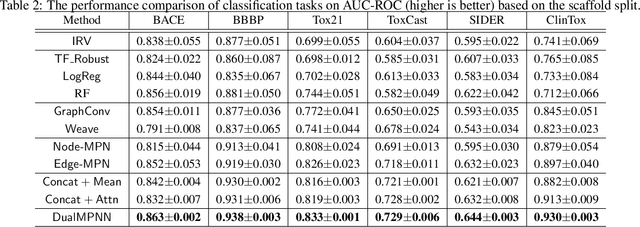
The crux of molecular property prediction is to generate meaningful representations of the molecules. One promising route is to exploit the molecular graph structure through Graph Neural Networks (GNNs). It is well known that both atoms and bonds significantly affect the chemical properties of a molecule, so an expressive model shall be able to exploit both node (atom) and edge (bond) information simultaneously. Guided by this observation, we present Multi-View Graph Neural Network (MV-GNN), a multi-view message passing architecture to enable more accurate predictions of molecular properties. In MV-GNN, we introduce a shared self-attentive readout component and disagreement loss to stabilize the training process. This readout component also renders the whole architecture interpretable. We further boost the expressive power of MV-GNN by proposing a cross-dependent message passing scheme that enhances information communication of the two views, which results in the MV-GNN^cross variant. Lastly, we theoretically justify the expressiveness of the two proposed models in terms of distinguishing non-isomorphism graphs. Extensive experiments demonstrate that MV-GNN models achieve remarkably superior performance over the state-of-the-art models on a variety of challenging benchmarks. Meanwhile, visualization results of the node importance are consistent with prior knowledge, which confirms the interpretability power of MV-GNN models.