 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTrillion Parameter AI Serving Infrastructure for Scientific Discovery: A Survey and Vision
Feb 05, 2024


Deep learning methods are transforming research, enabling new techniques, and ultimately leading to new discoveries. As the demand for more capable AI models continues to grow, we are now entering an era of Trillion Parameter Models (TPM), or models with more than a trillion parameters -- such as Huawei's PanGu-$\Sigma$. We describe a vision for the ecosystem of TPM users and providers that caters to the specific needs of the scientific community. We then outline the significant technical challenges and open problems in system design for serving TPMs to enable scientific research and discovery. Specifically, we describe the requirements of a comprehensive software stack and interfaces to support the diverse and flexible requirements of researchers.
WordScape: a Pipeline to extract multilingual, visually rich Documents with Layout Annotations from Web Crawl Data
Dec 15, 2023



We introduce WordScape, a novel pipeline for the creation of cross-disciplinary, multilingual corpora comprising millions of pages with annotations for document layout detection. Relating visual and textual items on document pages has gained further significance with the advent of multimodal models. Various approaches proved effective for visual question answering or layout segmentation. However, the interplay of text, tables, and visuals remains challenging for a variety of document understanding tasks. In particular, many models fail to generalize well to diverse domains and new languages due to insufficient availability of training data. WordScape addresses these limitations. Our automatic annotation pipeline parses the Open XML structure of Word documents obtained from the web, jointly providing layout-annotated document images and their textual representations. In turn, WordScape offers unique properties as it (1) leverages the ubiquity of the Word file format on the internet, (2) is readily accessible through the Common Crawl web corpus, (3) is adaptive to domain-specific documents, and (4) offers culturally and linguistically diverse document pages with natural semantic structure and high-quality text. Together with the pipeline, we will additionally release 9.5M urls to word documents which can be processed using WordScape to create a dataset of over 40M pages. Finally, we investigate the quality of text and layout annotations extracted by WordScape, assess the impact on document understanding benchmarks, and demonstrate that manual labeling costs can be substantially reduced.
DeepSpeed4Science Initiative: Enabling Large-Scale Scientific Discovery through Sophisticated AI System Technologies
Oct 11, 2023



In the upcoming decade, deep learning may revolutionize the natural sciences, enhancing our capacity to model and predict natural occurrences. This could herald a new era of scientific exploration, bringing significant advancements across sectors from drug development to renewable energy. To answer this call, we present DeepSpeed4Science initiative (deepspeed4science.ai) which aims to build unique capabilities through AI system technology innovations to help domain experts to unlock today's biggest science mysteries. By leveraging DeepSpeed's current technology pillars (training, inference and compression) as base technology enablers, DeepSpeed4Science will create a new set of AI system technologies tailored for accelerating scientific discoveries by addressing their unique complexity beyond the common technical approaches used for accelerating generic large language models (LLMs). In this paper, we showcase the early progress we made with DeepSpeed4Science in addressing two of the critical system challenges in structural biology research.
Towards a Modular Architecture for Science Factories
Aug 18, 2023Advances in robotic automation, high-performance computing (HPC), and artificial intelligence (AI) encourage us to conceive of science factories: large, general-purpose computation- and AI-enabled self-driving laboratories (SDLs) with the generality and scale needed both to tackle large discovery problems and to support thousands of scientists. Science factories require modular hardware and software that can be replicated for scale and (re)configured to support many applications. To this end, we propose a prototype modular science factory architecture in which reconfigurable modules encapsulating scientific instruments are linked with manipulators to form workcells, that can themselves be combined to form larger assemblages, and linked with distributed computing for simulation, AI model training and inference, and related tasks. Workflows that perform sets of actions on modules can be specified, and various applications, comprising workflows plus associated computational and data manipulation steps, can be run concurrently. We report on our experiences prototyping this architecture and applying it in experiments involving 15 different robotic apparatus, five applications (one in education, two in biology, two in materials), and a variety of workflows, across four laboratories. We describe the reuse of modules, workcells, and workflows in different applications, the migration of applications between workcells, and the use of digital twins, and suggest directions for future work aimed at yet more generality and scalability. Code and data are available at https://ad-sdl.github.io/wei2023 and in the Supplementary Information
Learning from learning machines: a new generation of AI technology to meet the needs of science
Nov 27, 2021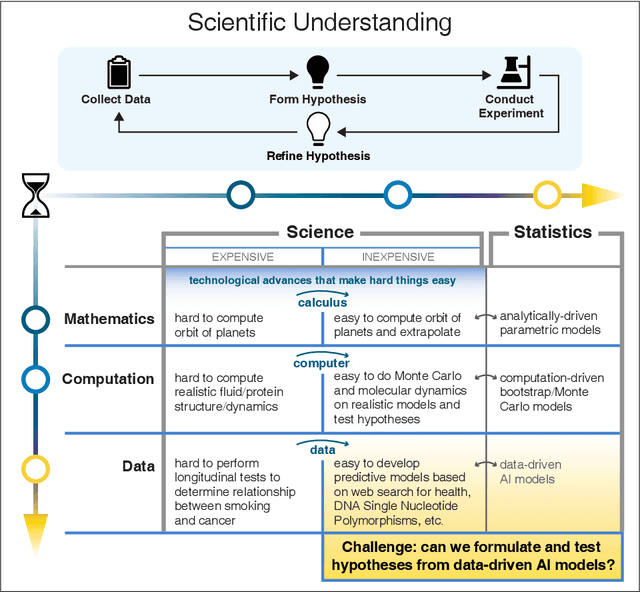

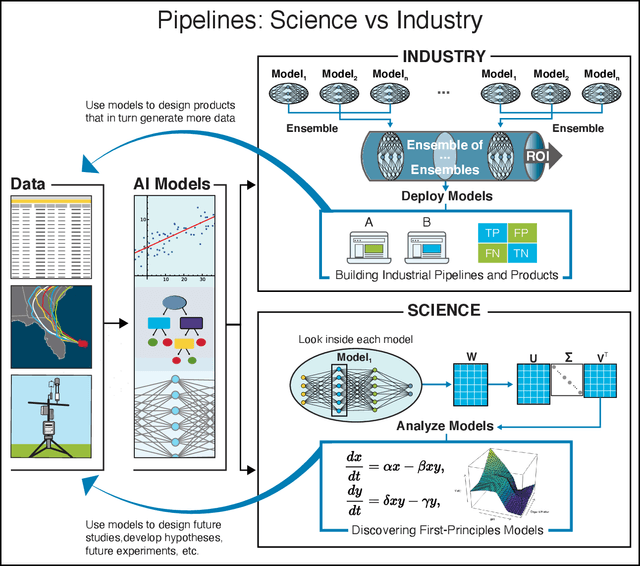
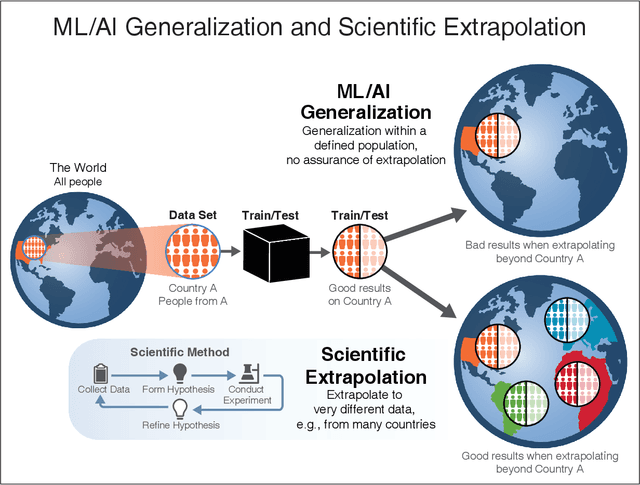
We outline emerging opportunities and challenges to enhance the utility of AI for scientific discovery. The distinct goals of AI for industry versus the goals of AI for science create tension between identifying patterns in data versus discovering patterns in the world from data. If we address the fundamental challenges associated with "bridging the gap" between domain-driven scientific models and data-driven AI learning machines, then we expect that these AI models can transform hypothesis generation, scientific discovery, and the scientific process itself.
Protein-Ligand Docking Surrogate Models: A SARS-CoV-2 Benchmark for Deep Learning Accelerated Virtual Screening
Jun 30, 2021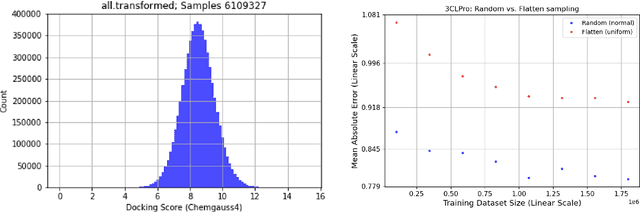

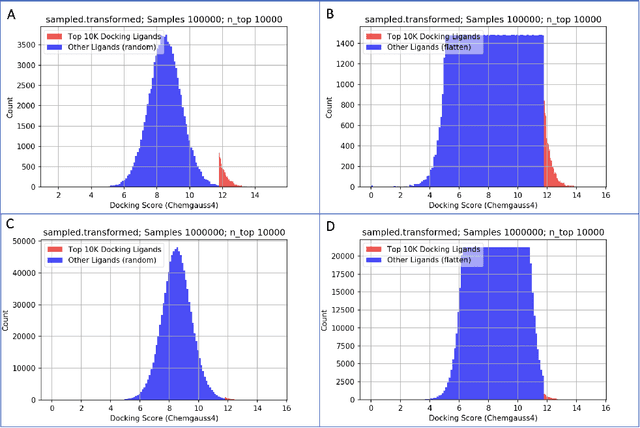
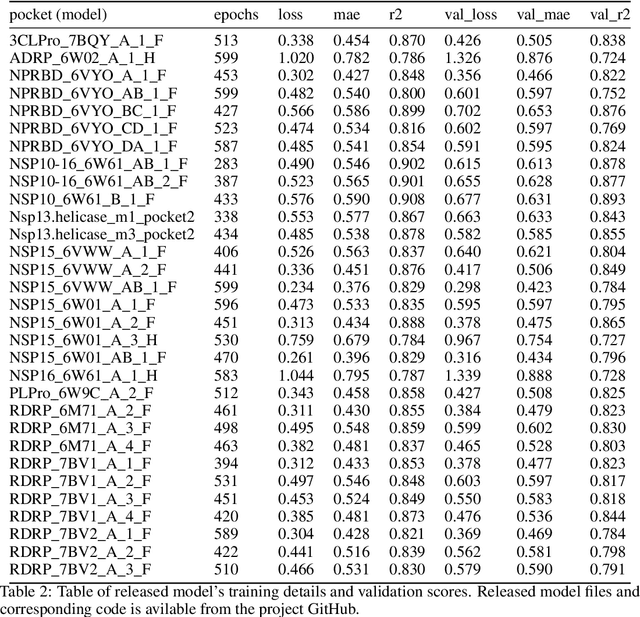
We propose a benchmark to study surrogate model accuracy for protein-ligand docking. We share a dataset consisting of 200 million 3D complex structures and 2D structure scores across a consistent set of 13 million "in-stock" molecules over 15 receptors, or binding sites, across the SARS-CoV-2 proteome. Our work shows surrogate docking models have six orders of magnitude more throughput than standard docking protocols on the same supercomputer node types. We demonstrate the power of high-speed surrogate models by running each target against 1 billion molecules in under a day (50k predictions per GPU seconds). We showcase a workflow for docking utilizing surrogate ML models as a pre-filter. Our workflow is ten times faster at screening a library of compounds than the standard technique, with an error rate less than 0.01\% of detecting the underlying best scoring 0.1\% of compounds. Our analysis of the speedup explains that to screen more molecules under a docking paradigm, another order of magnitude speedup must come from model accuracy rather than computing speed (which, if increased, will not anymore alter our throughput to screen molecules). We believe this is strong evidence for the community to begin focusing on improving the accuracy of surrogate models to improve the ability to screen massive compound libraries 100x or even 1000x faster than current techniques.
Neko: a Library for Exploring Neuromorphic Learning Rules
May 01, 2021
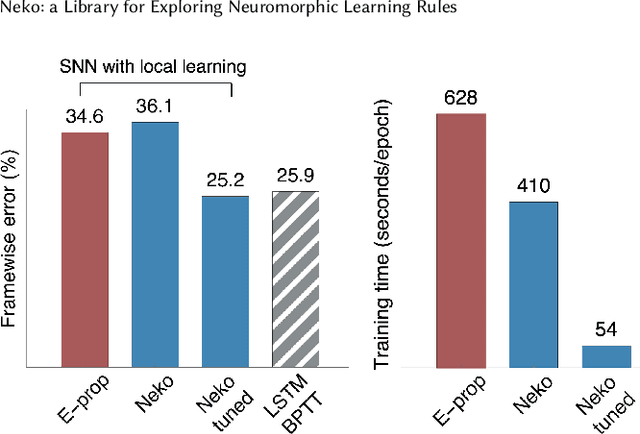
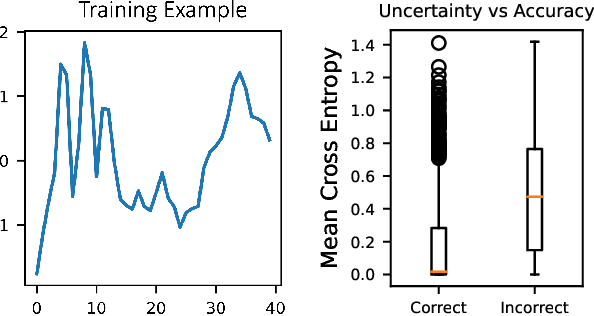
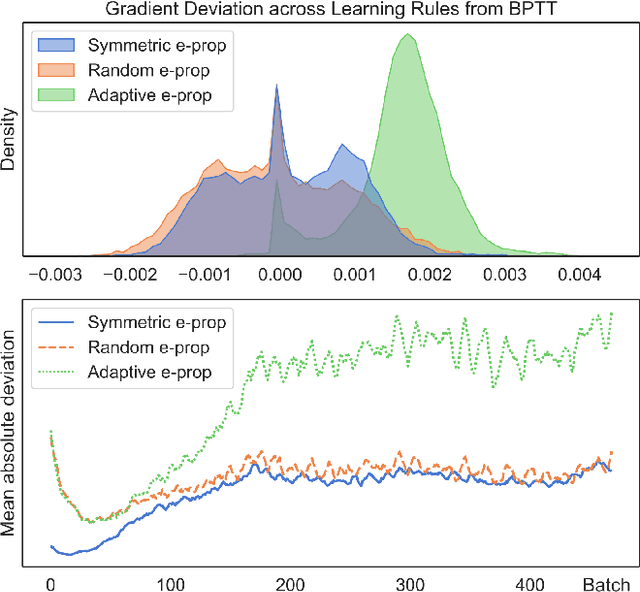
The field of neuromorphic computing is in a period of active exploration. While many tools have been developed to simulate neuronal dynamics or convert deep networks to spiking models, general software libraries for learning rules remain underexplored. This is partly due to the diverse, challenging nature of efforts to design new learning rules, which range from encoding methods to gradient approximations, from population approaches that mimic the Bayesian brain to constrained learning algorithms deployed on memristor crossbars. To address this gap, we present Neko, a modular, extensible library with a focus on aiding the design of new learning algorithms. We demonstrate the utility of Neko in three exemplar cases: online local learning, probabilistic learning, and analog on-device learning. Our results show that Neko can replicate the state-of-the-art algorithms and, in one case, lead to significant outperformance in accuracy and speed. Further, it offers tools including gradient comparison that can help develop new algorithmic variants. Neko is an open source Python library that supports PyTorch and TensorFlow backends.
Scaffold Embeddings: Learning the Structure Spanned by Chemical Fragments, Scaffolds and Compounds
Mar 11, 2021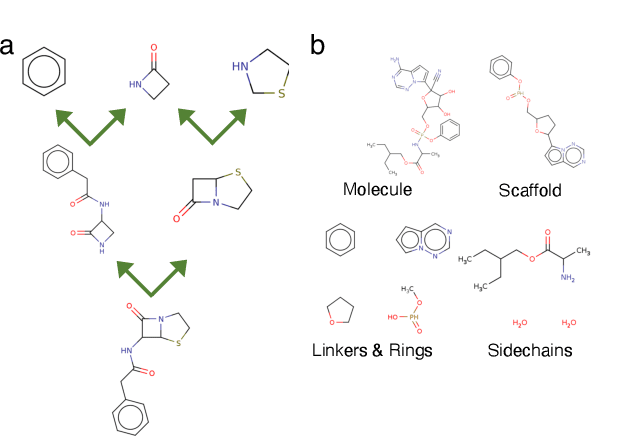

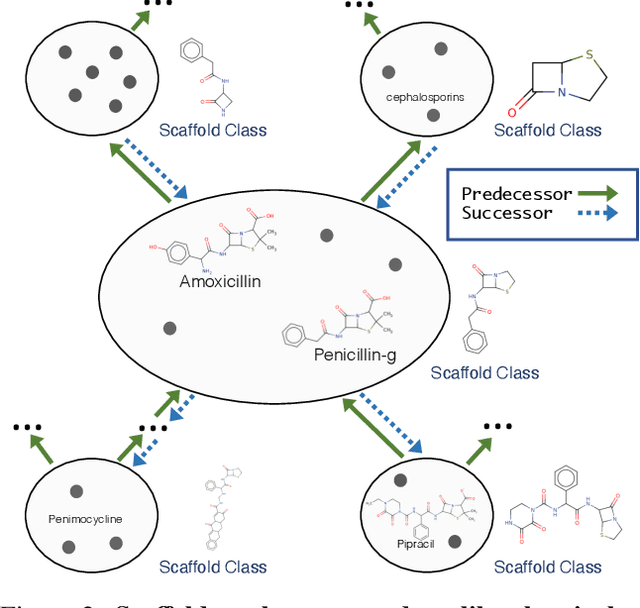
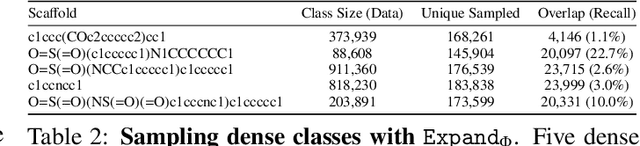
Molecules have seemed like a natural fit to deep learning's tendency to handle a complex structure through representation learning, given enough data. However, this often continuous representation is not natural for understanding chemical space as a domain and is particular to samples and their differences. We focus on exploring a natural structure for representing chemical space as a structured domain: embedding drug-like chemical space into an enumerable hypergraph based on scaffold classes linked through an inclusion operator. This paper shows how molecules form classes of scaffolds, how scaffolds relate to each in a hypergraph, and how this structure of scaffolds is natural for drug discovery workflows such as predicting properties and optimizing molecular structures. We compare the assumptions and utility of various embeddings of molecules, such as their respective induced distance metrics, their extendibility to represent chemical space as a structured domain, and the consequences of utilizing the structure for learning tasks.
Pandemic Drugs at Pandemic Speed: Accelerating COVID-19 Drug Discovery with Hybrid Machine Learning- and Physics-based Simulations on High Performance Computers
Mar 04, 2021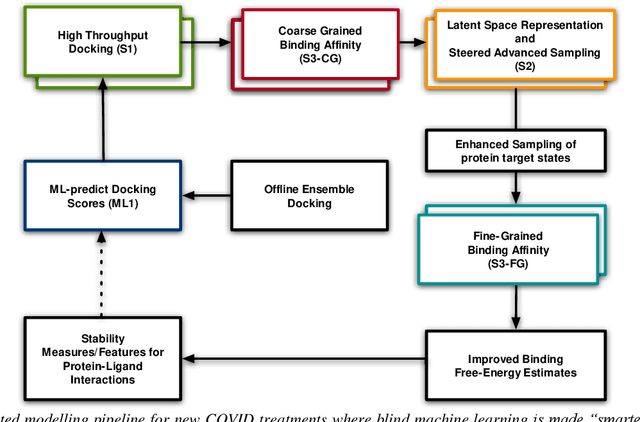

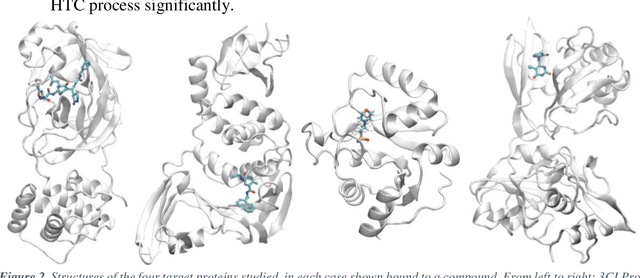
The race to meet the challenges of the global pandemic has served as a reminder that the existing drug discovery process is expensive, inefficient and slow. There is a major bottleneck screening the vast number of potential small molecules to shortlist lead compounds for antiviral drug development. New opportunities to accelerate drug discovery lie at the interface between machine learning methods, in this case developed for linear accelerators, and physics-based methods. The two in silico methods, each have their own advantages and limitations which, interestingly, complement each other. Here, we present an innovative method that combines both approaches to accelerate drug discovery. The scale of the resulting workflow is such that it is dependent on high performance computing. We have demonstrated the applicability of this workflow on four COVID-19 target proteins and our ability to perform the required large-scale calculations to identify lead compounds on a variety of supercomputers.
Learning Curves for Drug Response Prediction in Cancer Cell Lines
Nov 25, 2020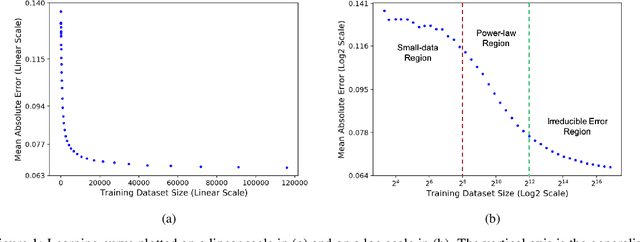
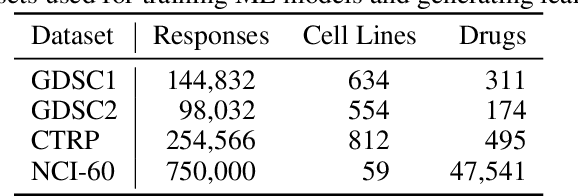
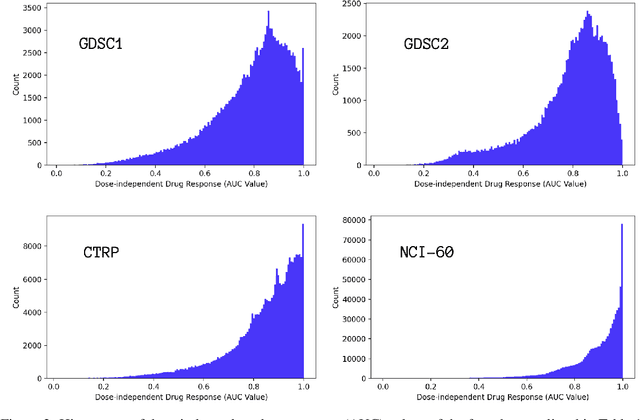
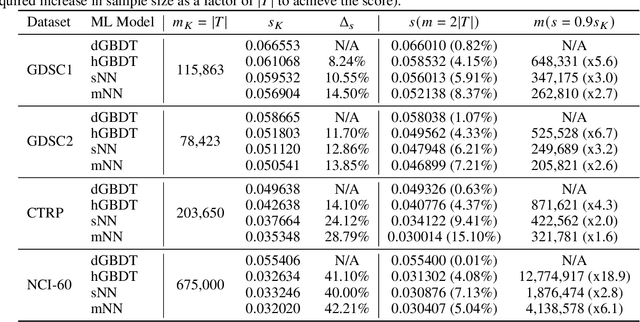
Motivated by the size of cell line drug sensitivity data, researchers have been developing machine learning (ML) models for predicting drug response to advance cancer treatment. As drug sensitivity studies continue generating data, a common question is whether the proposed predictors can further improve the generalization performance with more training data. We utilize empirical learning curves for evaluating and comparing the data scaling properties of two neural networks (NNs) and two gradient boosting decision tree (GBDT) models trained on four drug screening datasets. The learning curves are accurately fitted to a power law model, providing a framework for assessing the data scaling behavior of these predictors. The curves demonstrate that no single model dominates in terms of prediction performance across all datasets and training sizes, suggesting that the shape of these curves depends on the unique model-dataset pair. The multi-input NN (mNN), in which gene expressions and molecular drug descriptors are input into separate subnetworks, outperforms a single-input NN (sNN), where the cell and drug features are concatenated for the input layer. In contrast, a GBDT with hyperparameter tuning exhibits superior performance as compared with both NNs at the lower range of training sizes for two of the datasets, whereas the mNN performs better at the higher range of training sizes. Moreover, the trajectory of the curves suggests that increasing the sample size is expected to further improve prediction scores of both NNs. These observations demonstrate the benefit of using learning curves to evaluate predictors, providing a broader perspective on the overall data scaling characteristics. The fitted power law curves provide a forward-looking performance metric and can serve as a co-design tool to guide experimental biologists and computational scientists in the design of future experiments.