 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEEG based Emotion Recognition: A Tutorial and Review
Mar 16, 2022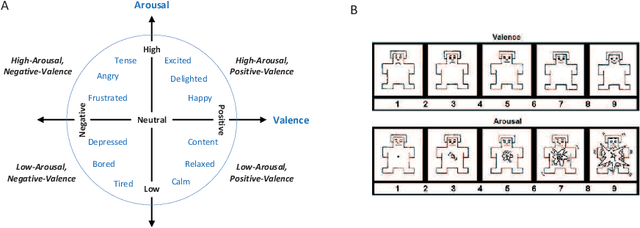
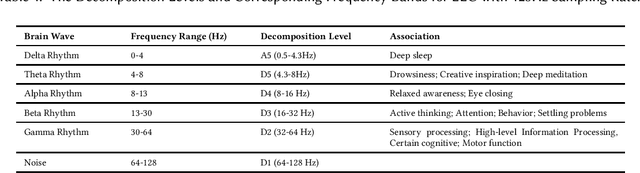
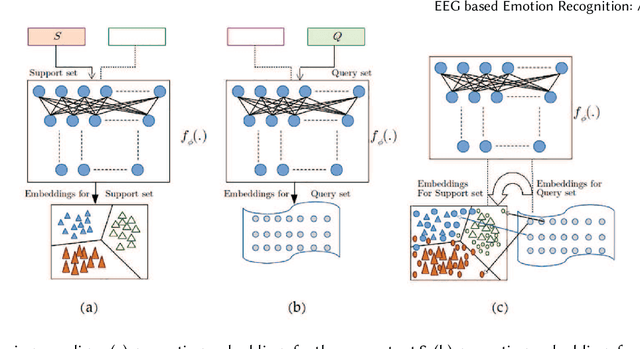
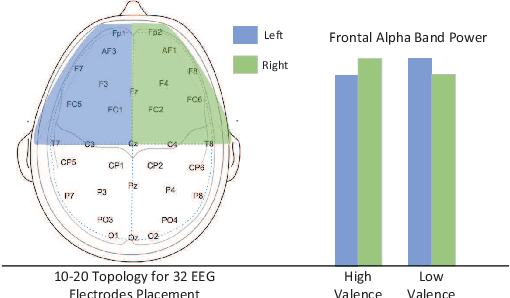
Emotion recognition technology through analyzing the EEG signal is currently an essential concept in Artificial Intelligence and holds great potential in emotional health care, human-computer interaction, multimedia content recommendation, etc. Though there have been several works devoted to reviewing EEG-based emotion recognition, the content of these reviews needs to be updated. In addition, those works are either fragmented in content or only focus on specific techniques adopted in this area but neglect the holistic perspective of the entire technical routes. Hence, in this paper, we review from the perspective of researchers who try to take the first step on this topic. We review the recent representative works in the EEG-based emotion recognition research and provide a tutorial to guide the researchers to start from the beginning. The scientific basis of EEG-based emotion recognition in the psychological and physiological levels is introduced. Further, we categorize these reviewed works into different technical routes and illustrate the theoretical basis and the research motivation, which will help the readers better understand why those techniques are studied and employed. At last, existing challenges and future investigations are also discussed in this paper, which guides the researchers to decide potential future research directions.
Deconfounded Representation Similarity for Comparison of Neural Networks
Jan 31, 2022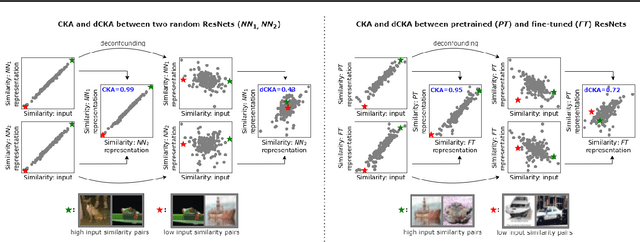
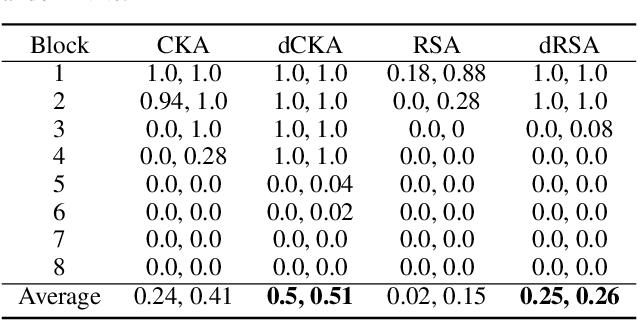
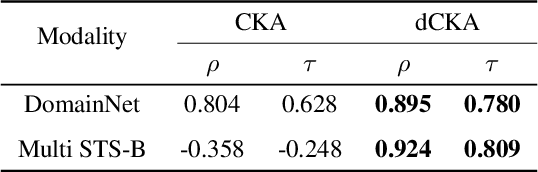
Similarity metrics such as representational similarity analysis (RSA) and centered kernel alignment (CKA) have been used to compare layer-wise representations between neural networks. However, these metrics are confounded by the population structure of data items in the input space, leading to spuriously high similarity for even completely random neural networks and inconsistent domain relations in transfer learning. We introduce a simple and generally applicable fix to adjust for the confounder with covariate adjustment regression, which retains the intuitive invariance properties of the original similarity measures. We show that deconfounding the similarity metrics increases the resolution of detecting semantically similar neural networks. Moreover, in real-world applications, deconfounding improves the consistency of representation similarities with domain similarities in transfer learning, and increases correlation with out-of-distribution accuracy.
A Unified Review of Deep Learning for Automated Medical Coding
Jan 08, 2022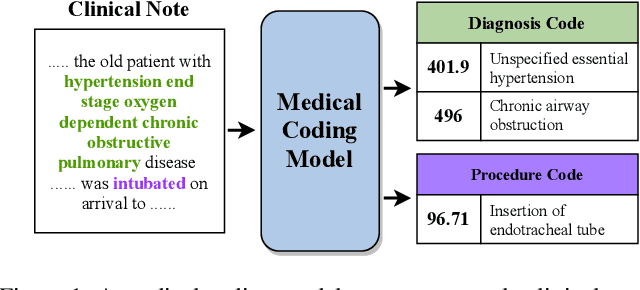
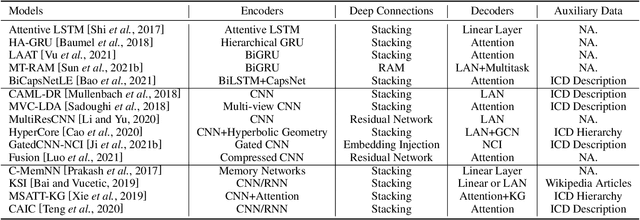
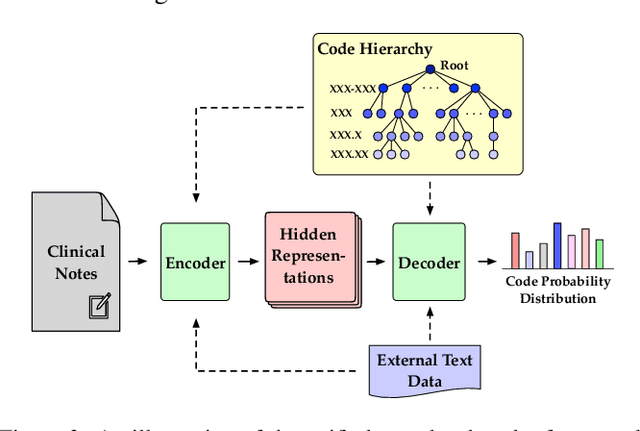
Automated medical coding, an essential task for healthcare operation and delivery, makes unstructured data manageable by predicting medical codes from clinical documents. Recent advances in deep learning models in natural language processing have been widely applied to this task. However, it lacks a unified view of the design of neural network architectures for medical coding. This review proposes a unified framework to provide a general understanding of the building blocks of medical coding models and summarizes recent advanced models under the proposed framework. Our unified framework decomposes medical coding into four main components, i.e., encoder modules for text feature extraction, mechanisms for building deep encoder architectures, decoder modules for transforming hidden representations into medical codes, and the usage of auxiliary information. Finally, we discuss key research challenges and future directions.
Patient Outcome and Zero-shot Diagnosis Prediction with Hypernetwork-guided Multitask Learning
Sep 07, 2021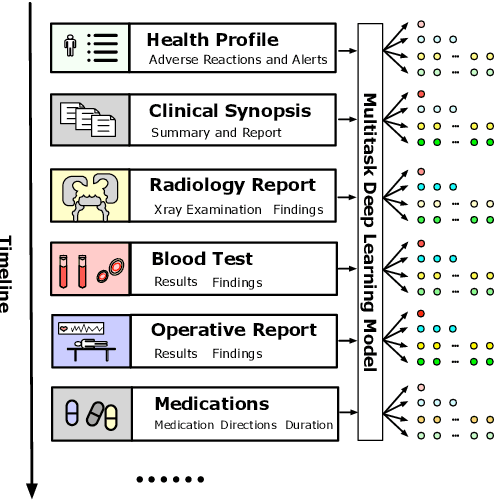

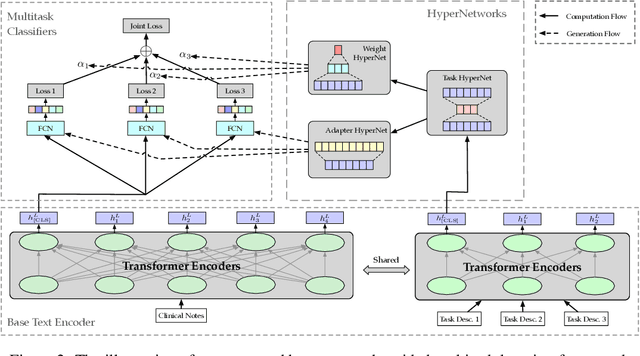
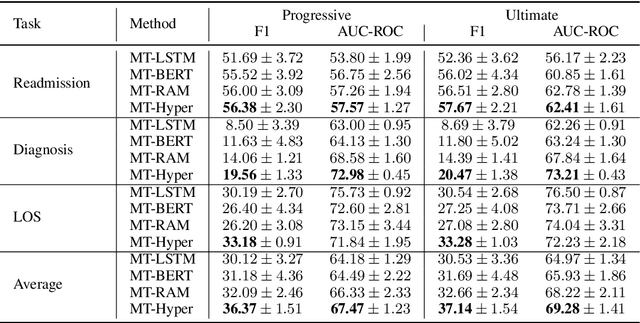
Multitask deep learning has been applied to patient outcome prediction from text, taking clinical notes as input and training deep neural networks with a joint loss function of multiple tasks. However, the joint training scheme of multitask learning suffers from inter-task interference, and diagnosis prediction among the multiple tasks has the generalizability issue due to rare diseases or unseen diagnoses. To solve these challenges, we propose a hypernetwork-based approach that generates task-conditioned parameters and coefficients of multitask prediction heads to learn task-specific prediction and balance the multitask learning. We also incorporate semantic task information to improves the generalizability of our task-conditioned multitask model. Experiments on early and discharge notes extracted from the real-world MIMIC database show our method can achieve better performance on multitask patient outcome prediction than strong baselines in most cases. Besides, our method can effectively handle the scenario with limited information and improve zero-shot prediction on unseen diagnosis categories.
Multi-task Balanced and Recalibrated Network for Medical Code Prediction
Sep 06, 2021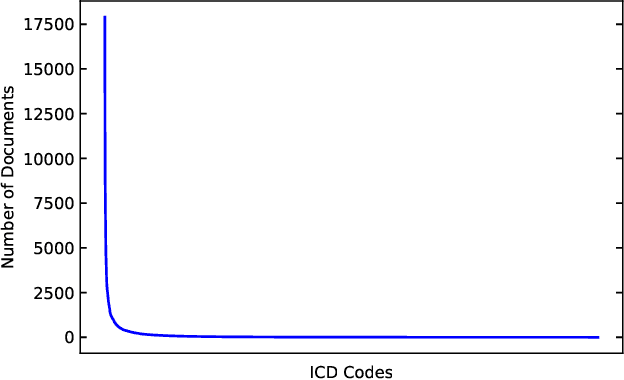
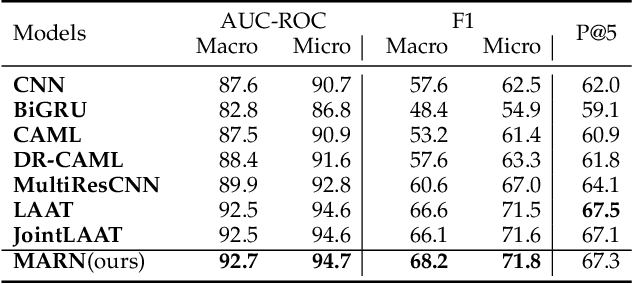
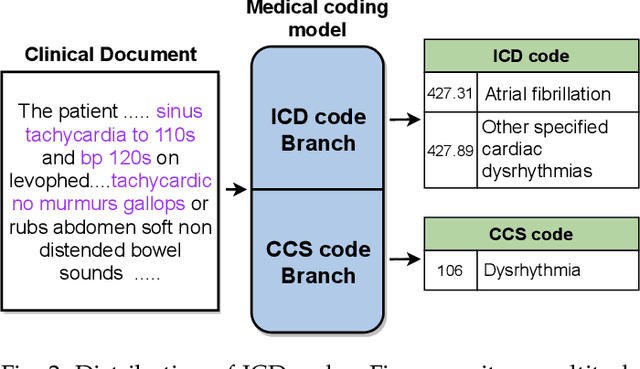
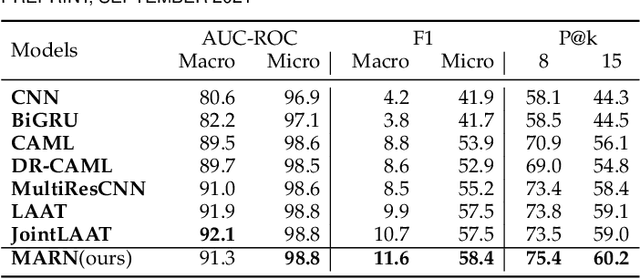
Human coders assign standardized medical codes to clinical documents generated during patients' hospitalization, which is error-prone and labor-intensive. Automated medical coding approaches have been developed using machine learning methods such as deep neural networks. Nevertheless, automated medical coding is still challenging because of the imbalanced class problem, complex code association, and noise in lengthy documents. To solve these difficulties, we propose a novel neural network called Multi-task Balanced and Recalibrated Neural Network. Significantly, the multi-task learning scheme shares the relationship knowledge between different code branches to capture the code association. A recalibrated aggregation module is developed by cascading convolutional blocks to extract high-level semantic features that mitigate the impact of noise in documents. Also, the cascaded structure of the recalibrated module can benefit the learning from lengthy notes. To solve the class imbalanced problem, we deploy the focal loss to redistribute the attention of low and high-frequency medical codes. Experimental results show that our proposed model outperforms competitive baselines on a real-world clinical dataset MIMIC-III.
Medical SANSformers: Training self-supervised transformers without attention for Electronic Medical Records
Aug 31, 2021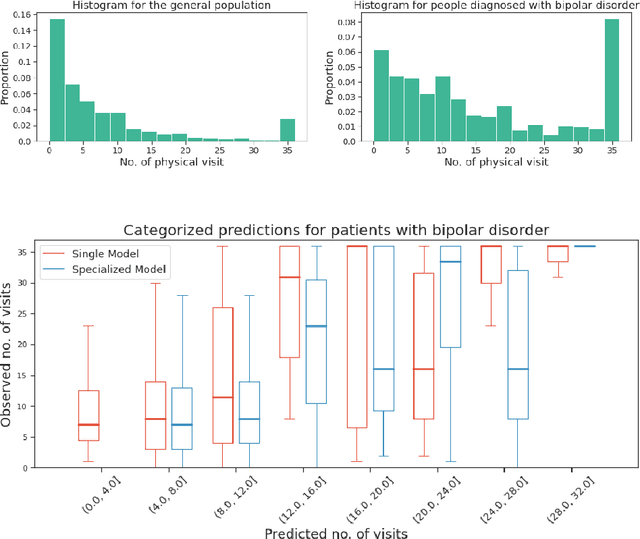
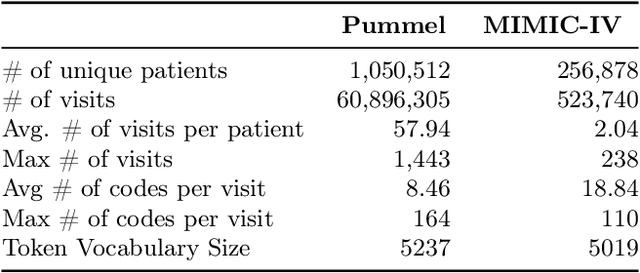
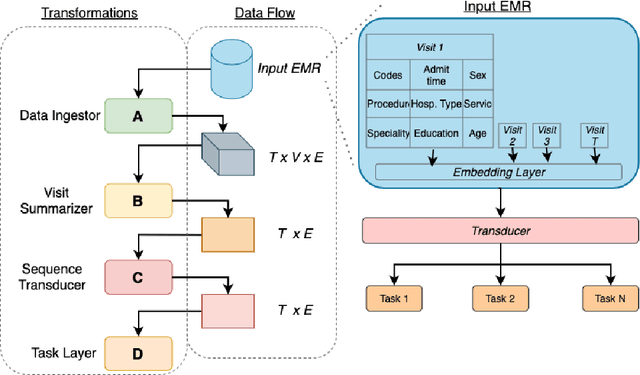
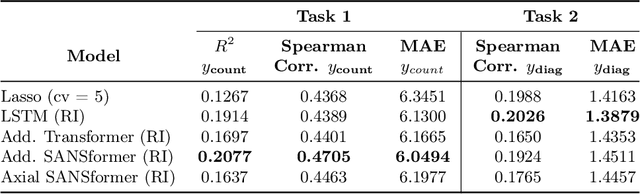
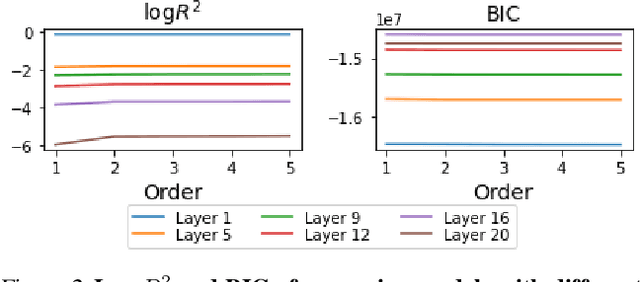
We leverage deep sequential models to tackle the problem of predicting healthcare utilization for patients, which could help governments to better allocate resources for future healthcare use. Specifically, we study the problem of \textit{divergent subgroups}, wherein the outcome distribution in a smaller subset of the population considerably deviates from that of the general population. The traditional approach for building specialized models for divergent subgroups could be problematic if the size of the subgroup is very small (for example, rare diseases). To address this challenge, we first develop a novel attention-free sequential model, SANSformers, instilled with inductive biases suited for modeling clinical codes in electronic medical records. We then design a task-specific self-supervision objective and demonstrate its effectiveness, particularly in scarce data settings, by pre-training each model on the entire health registry (with close to one million patients) before fine-tuning for downstream tasks on the divergent subgroups. We compare the novel SANSformer architecture with the LSTM and Transformer models using two data sources and a multi-task learning objective that aids healthcare utilization prediction. Empirically, the attention-free SANSformer models perform consistently well across experiments, outperforming the baselines in most cases by at least $\sim 10$\%. Furthermore, the self-supervised pre-training boosts performance significantly throughout, for example by over $\sim 50$\% (and as high as $800$\%) on $R^2$ score when predicting the number of hospital visits.
Deep Learning for Depression Recognition with Audiovisual Cues: A Review
May 27, 2021With the acceleration of the pace of work and life, people have to face more and more pressure, which increases the possibility of suffering from depression. However, many patients may fail to get a timely diagnosis due to the serious imbalance in the doctor-patient ratio in the world. Promisingly, physiological and psychological studies have indicated some differences in speech and facial expression between patients with depression and healthy individuals. Consequently, to improve current medical care, many scholars have used deep learning to extract a representation of depression cues in audio and video for automatic depression detection. To sort out and summarize these works, this review introduces the databases and describes objective markers for automatic depression estimation (ADE). Furthermore, we review the deep learning methods for automatic depression detection to extract the representation of depression from audio and video. Finally, this paper discusses challenges and promising directions related to automatic diagnosing of depression using deep learning technologies.
Multitask Recalibrated Aggregation Network for Medical Code Prediction
Apr 30, 2021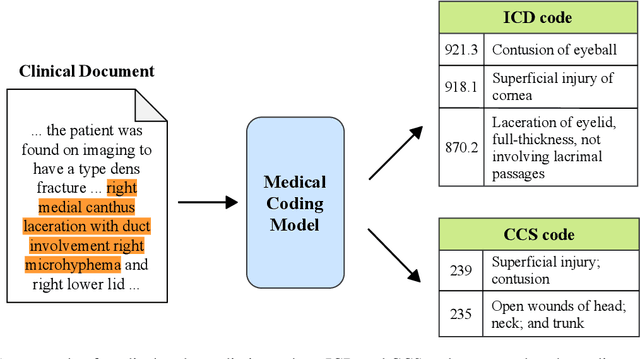
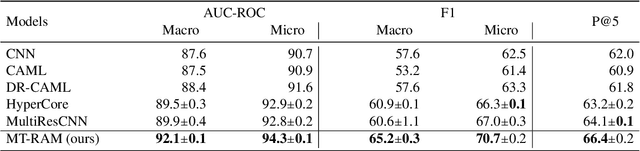
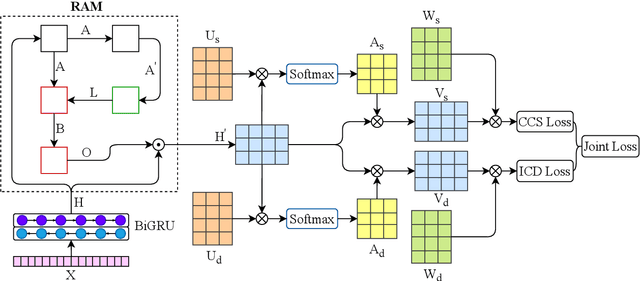
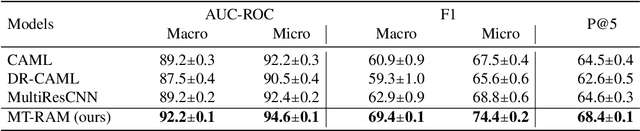
Medical coding translates professionally written medical reports into standardized codes, which is an essential part of medical information systems and health insurance reimbursement. Manual coding by trained human coders is time-consuming and error-prone. Thus, automated coding algorithms have been developed, building especially on the recent advances in machine learning and deep neural networks. To solve the challenges of encoding lengthy and noisy clinical documents and capturing code associations, we propose a multitask recalibrated aggregation network. In particular, multitask learning shares information across different coding schemes and captures the dependencies between different medical codes. Feature recalibration and aggregation in shared modules enhance representation learning for lengthy notes. Experiments with a real-world MIMIC-III dataset show significantly improved predictive performance.
A Critical Look At The Identifiability of Causal Effects with Deep Latent Variable Models
Mar 16, 2021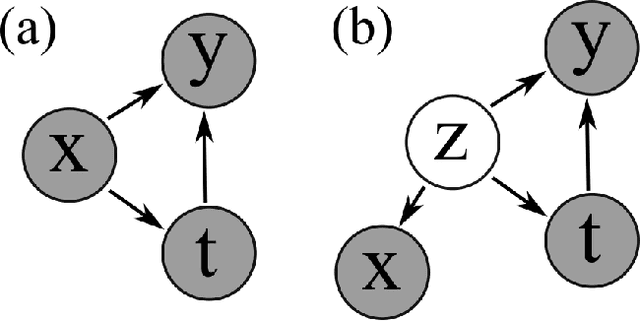
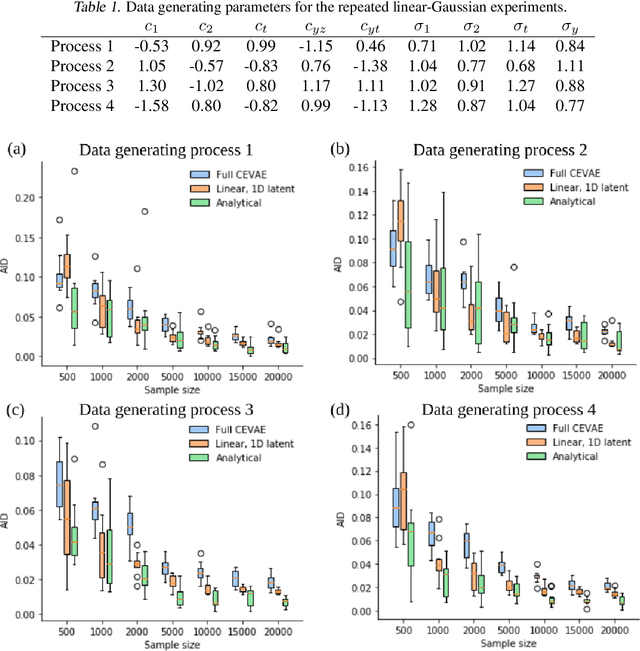
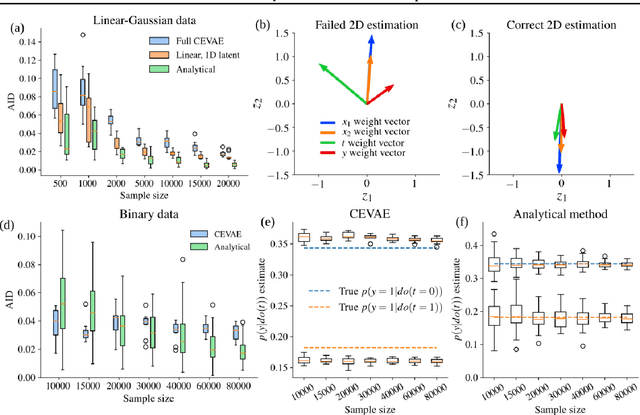
Using deep latent variable models in causal inference has attracted considerable interest recently, but an essential open question is their identifiability. While they have yielded promising results and theory exists on the identifiability of some simple model formulations, we also know that causal effects cannot be identified in general with latent variables. We investigate this gap between theory and empirical results with theoretical considerations and extensive experiments under multiple synthetic and real-world data sets, using the causal effect variational autoencoder (CEVAE) as a case study. While CEVAE seems to work reliably under some simple scenarios, it does not identify the correct causal effect with a misspecified latent variable or a complex data distribution, as opposed to the original goals of the model. Our results show that the question of identifiability cannot be disregarded, and we argue that more attention should be paid to it in future work.
Does the Magic of BERT Apply to Medical Code Assignment? A Quantitative Study
Mar 11, 2021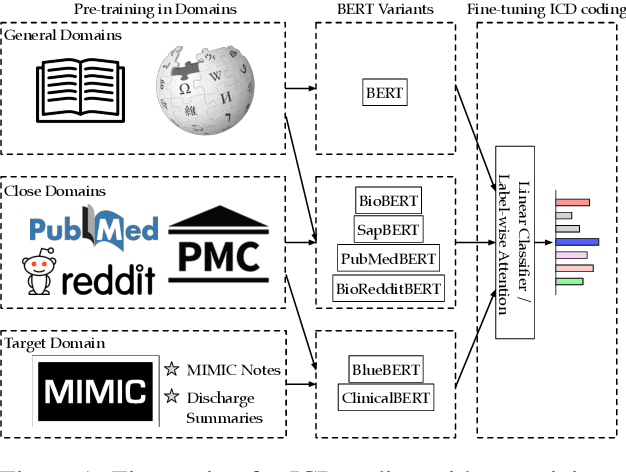
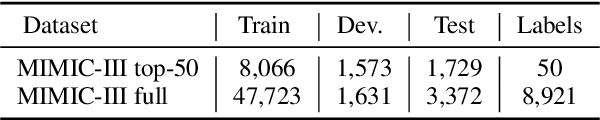
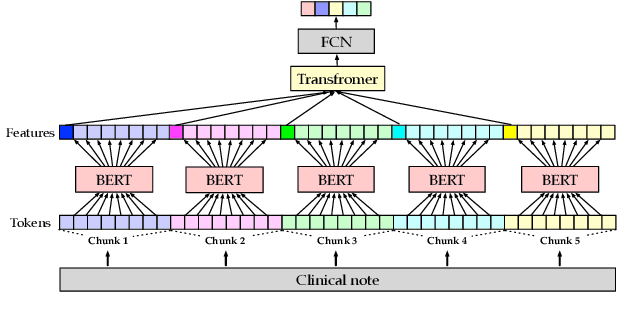
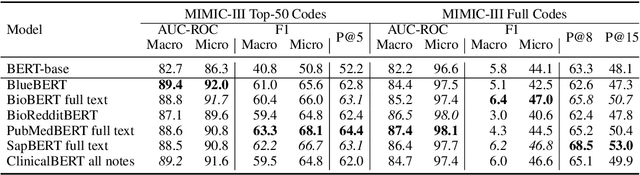
Unsupervised pretraining is an integral part of many natural language processing systems, and transfer learning with language models has achieved remarkable results in many downstream tasks. In the clinical application of medical code assignment, diagnosis and procedure codes are inferred from lengthy clinical notes such as hospital discharge summaries. However, it is not clear if pretrained models are useful for medical code prediction without further architecture engineering. This paper conducts a comprehensive quantitative analysis of various contextualized language models' performance, pretrained in different domains, for medical code assignment from clinical notes. We propose a hierarchical fine-tuning architecture to capture interactions between distant words and adopt label-wise attention to exploit label information. Contrary to current trends, we demonstrate that a carefully trained classical CNN outperforms attention-based models on a MIMIC-III subset with frequent codes. Our empirical findings suggest directions for improving the medical code assignment application.