 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePrompt Codebooks: Discrete Compositional Optimization for Language Model Instruction Refinement
May 27, 2026Automatic prompt optimization (APO) has driven significant gains in LLM-based agentic workflows. However, existing methods treat each task's prompt as a monolithic, instance-blind string optimized through global edits, producing brittle updates and preventing the reuse of learned sub-behaviors. We propose Prompt Codebooks (PCO), a novel compositional prompt optimization framework that recasts APO as discrete learning over a finite vocabulary of natural-language instincts - atomic, reusable instruction units. PCO organizes prompt-construction knowledge in a discrete codebook and routes each input to a small subset of entries via an LLM-based encoder; a generator composes them into a prompt for the frozen target model; a critic emits a structured verdict that decomposes by attribution into per-variable textual gradients, jointly training the encoder, generator, and codebook under a language-valued min-max objective. The resulting routing is per-instance: different inputs in the same task receive different instinct compositions, a regime structurally inexpressible under instance-blind methods. Across six benchmarks on Qwen3-8B and LLaMA-3.1-8B, PCO improves over zero-shot by up to +30.36 points, surpasses the strongest prior baseline (GEPA) by +3.34 on HotpotQA and +1.11 in aggregate, and reduces deployed prompt length by up to 14.1x versus MIPROv2 and 3.0x versus GEPA using only K=16 instincts.
GOLDMARK: Governed Outcome-Linked Diagnostic Model Assessment Reference Kit
Mar 21, 2026Computational biomarkers (CBs) are histopathology-derived patterns extracted from hematoxylin-eosin (H&E) whole-slide images (WSIs) using artificial intelligence (AI) to predict therapeutic response or prognosis. Recently, slide-level multiple-instance learning (MIL) with pathology foundation models (PFMs) has become the standard baseline for CB development. While these methods have improved predictive performance, computational pathology lacks standardized intermediate data formats, provenance tracking, checkpointing conventions, and reproducible evaluation metrics required for clinical-grade deployment. We introduce GOLDMARK (https://artificialintelligencepathology.org), a standardized benchmarking framework built on a curated TCGA cohort with clinically actionable OncoKB level 1-3 biomarker labels. GOLDMARK releases structured intermediate representations, including tile coordinate maps, per-slide feature embeddings from canonical PFMs, quality-control metadata, predefined patient-level splits, trained slide-level models, and evaluation outputs. Models are trained on TCGA and evaluated on an independent MSKCC cohort with reciprocal testing. Across 33 tumor-biomarker tasks, mean AUROC was 0.689 (TCGA) and 0.630 (MSKCC). Restricting to the eight highest-performing tasks yielded mean AUROCs of 0.831 and 0.801, respectively. These tasks correspond to established morphologic-genomic associations (e.g., LGG IDH1, COAD MSI/BRAF, THCA BRAF/NRAS, BLCA FGFR3, UCEC PTEN) and showed the most stable cross-site performance. Differences between canonical encoders were modest relative to task-specific variability. GOLDMARK establishes a shared experimental substrate for computational pathology, enabling reproducible benchmarking and direct comparison of methods across datasets and models.
Single GPU Task Adaptation of Pathology Foundation Models for Whole Slide Image Analysis
Jun 05, 2025
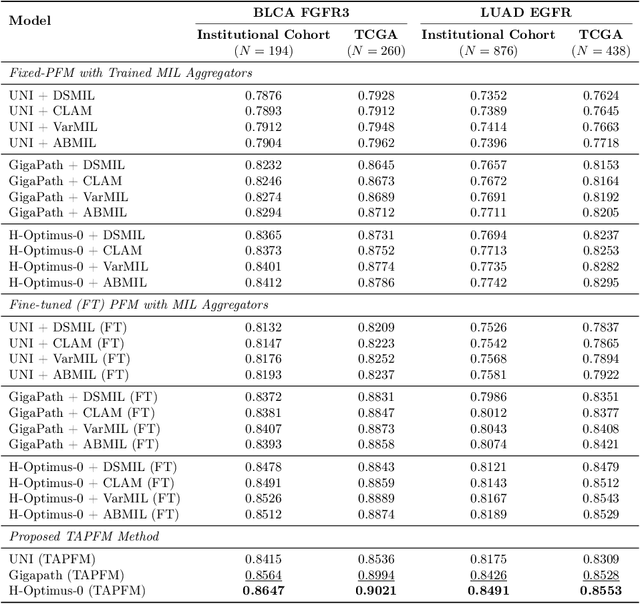
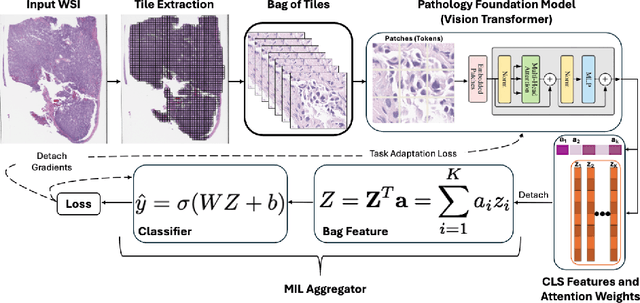
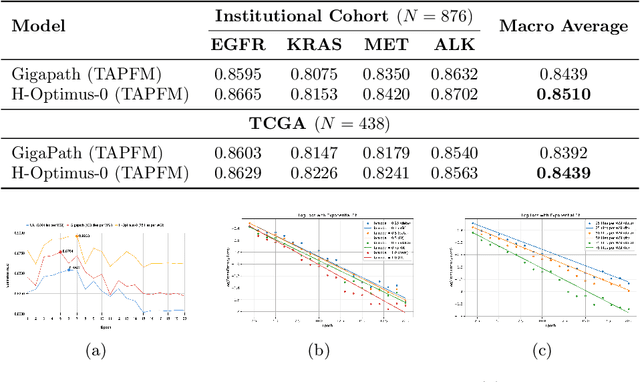
Pathology foundation models (PFMs) have emerged as powerful tools for analyzing whole slide images (WSIs). However, adapting these pretrained PFMs for specific clinical tasks presents considerable challenges, primarily due to the availability of only weak (WSI-level) labels for gigapixel images, necessitating multiple instance learning (MIL) paradigm for effective WSI analysis. This paper proposes a novel approach for single-GPU \textbf{T}ask \textbf{A}daptation of \textbf{PFM}s (TAPFM) that uses vision transformer (\vit) attention for MIL aggregation while optimizing both for feature representations and attention weights. The proposed approach maintains separate computational graphs for MIL aggregator and the PFM to create stable training dynamics that align with downstream task objectives during end-to-end adaptation. Evaluated on mutation prediction tasks for bladder cancer and lung adenocarcinoma across institutional and TCGA cohorts, TAPFM consistently outperforms conventional approaches, with H-Optimus-0 (TAPFM) outperforming the benchmarks. TAPFM effectively handles multi-label classification of actionable mutations as well. Thus, TAPFM makes adaptation of powerful pre-trained PFMs practical on standard hardware for various clinical applications.
Electrical Load Forecasting in Smart Grid: A Personalized Federated Learning Approach
Nov 15, 2024



Electric load forecasting is essential for power management and stability in smart grids. This is mainly achieved via advanced metering infrastructure, where smart meters (SMs) are used to record household energy consumption. Traditional machine learning (ML) methods are often employed for load forecasting but require data sharing which raises data privacy concerns. Federated learning (FL) can address this issue by running distributed ML models at local SMs without data exchange. However, current FL-based approaches struggle to achieve efficient load forecasting due to imbalanced data distribution across heterogeneous SMs. This paper presents a novel personalized federated learning (PFL) method to load prediction under non-independent and identically distributed (non-IID) metering data settings. Specifically, we introduce meta-learning, where the learning rates are manipulated using the meta-learning idea to maximize the gradient for each client in each global round. Clients with varying processing capacities, data sizes, and batch sizes can participate in global model aggregation and improve their local load forecasting via personalized learning. Simulation results show that our approach outperforms state-of-the-art ML and FL methods in terms of better load forecasting accuracy.
CACTUS: Chemistry Agent Connecting Tool-Usage to Science
May 02, 2024Large language models (LLMs) have shown remarkable potential in various domains, but they often lack the ability to access and reason over domain-specific knowledge and tools. In this paper, we introduced CACTUS (Chemistry Agent Connecting Tool-Usage to Science), an LLM-based agent that integrates cheminformatics tools to enable advanced reasoning and problem-solving in chemistry and molecular discovery. We evaluate the performance of CACTUS using a diverse set of open-source LLMs, including Gemma-7b, Falcon-7b, MPT-7b, Llama2-7b, and Mistral-7b, on a benchmark of thousands of chemistry questions. Our results demonstrate that CACTUS significantly outperforms baseline LLMs, with the Gemma-7b and Mistral-7b models achieving the highest accuracy regardless of the prompting strategy used. Moreover, we explore the impact of domain-specific prompting and hardware configurations on model performance, highlighting the importance of prompt engineering and the potential for deploying smaller models on consumer-grade hardware without significant loss in accuracy. By combining the cognitive capabilities of open-source LLMs with domain-specific tools, CACTUS can assist researchers in tasks such as molecular property prediction, similarity searching, and drug-likeness assessment. Furthermore, CACTUS represents a significant milestone in the field of cheminformatics, offering an adaptable tool for researchers engaged in chemistry and molecular discovery. By integrating the strengths of open-source LLMs with domain-specific tools, CACTUS has the potential to accelerate scientific advancement and unlock new frontiers in the exploration of novel, effective, and safe therapeutic candidates, catalysts, and materials. Moreover, CACTUS's ability to integrate with automated experimentation platforms and make data-driven decisions in real time opens up new possibilities for autonomous discovery.
Style Description based Text-to-Speech with Conditional Prosodic Layer Normalization based Diffusion GAN
Oct 27, 2023In this paper, we present a Diffusion GAN based approach (Prosodic Diff-TTS) to generate the corresponding high-fidelity speech based on the style description and content text as an input to generate speech samples within only 4 denoising steps. It leverages the novel conditional prosodic layer normalization to incorporate the style embeddings into the multi head attention based phoneme encoder and mel spectrogram decoder based generator architecture to generate the speech. The style embedding is generated by fine tuning the pretrained BERT model on auxiliary tasks such as pitch, speaking speed, emotion,gender classifications. We demonstrate the efficacy of our proposed architecture on multi-speaker LibriTTS and PromptSpeech datasets, using multiple quantitative metrics that measure generated accuracy and MOS.
Distraction-free Embeddings for Robust VQA
Aug 31, 2023



The generation of effective latent representations and their subsequent refinement to incorporate precise information is an essential prerequisite for Vision-Language Understanding (VLU) tasks such as Video Question Answering (VQA). However, most existing methods for VLU focus on sparsely sampling or fine-graining the input information (e.g., sampling a sparse set of frames or text tokens), or adding external knowledge. We present a novel "DRAX: Distraction Removal and Attended Cross-Alignment" method to rid our cross-modal representations of distractors in the latent space. We do not exclusively confine the perception of any input information from various modalities but instead use an attention-guided distraction removal method to increase focus on task-relevant information in latent embeddings. DRAX also ensures semantic alignment of embeddings during cross-modal fusions. We evaluate our approach on a challenging benchmark (SUTD-TrafficQA dataset), testing the framework's abilities for feature and event queries, temporal relation understanding, forecasting, hypothesis, and causal analysis through extensive experiments.
An Effective Meaningful Way to Evaluate Survival Models
Jun 01, 2023One straightforward metric to evaluate a survival prediction model is based on the Mean Absolute Error (MAE) -- the average of the absolute difference between the time predicted by the model and the true event time, over all subjects. Unfortunately, this is challenging because, in practice, the test set includes (right) censored individuals, meaning we do not know when a censored individual actually experienced the event. In this paper, we explore various metrics to estimate MAE for survival datasets that include (many) censored individuals. Moreover, we introduce a novel and effective approach for generating realistic semi-synthetic survival datasets to facilitate the evaluation of metrics. Our findings, based on the analysis of the semi-synthetic datasets, reveal that our proposed metric (MAE using pseudo-observations) is able to rank models accurately based on their performance, and often closely matches the true MAE -- in particular, is better than several alternative methods.
The ACROBAT 2022 Challenge: Automatic Registration Of Breast Cancer Tissue
May 29, 2023The alignment of tissue between histopathological whole-slide-images (WSI) is crucial for research and clinical applications. Advances in computing, deep learning, and availability of large WSI datasets have revolutionised WSI analysis. Therefore, the current state-of-the-art in WSI registration is unclear. To address this, we conducted the ACROBAT challenge, based on the largest WSI registration dataset to date, including 4,212 WSIs from 1,152 breast cancer patients. The challenge objective was to align WSIs of tissue that was stained with routine diagnostic immunohistochemistry to its H&E-stained counterpart. We compare the performance of eight WSI registration algorithms, including an investigation of the impact of different WSI properties and clinical covariates. We find that conceptually distinct WSI registration methods can lead to highly accurate registration performances and identify covariates that impact performances across methods. These results establish the current state-of-the-art in WSI registration and guide researchers in selecting and developing methods.
KL Regularized Normalization Framework for Low Resource Tasks
Dec 21, 2022



Large pre-trained models, such as Bert, GPT, and Wav2Vec, have demonstrated great potential for learning representations that are transferable to a wide variety of downstream tasks . It is difficult to obtain a large quantity of supervised data due to the limited availability of resources and time. In light of this, a significant amount of research has been conducted in the area of adopting large pre-trained datasets for diverse downstream tasks via fine tuning, linear probing, or prompt tuning in low resource settings. Normalization techniques are essential for accelerating training and improving the generalization of deep neural networks and have been successfully used in a wide variety of applications. A lot of normalization techniques have been proposed but the success of normalization in low resource downstream NLP and speech tasks is limited. One of the reasons is the inability to capture expressiveness by rescaling parameters of normalization. We propose KullbackLeibler(KL) Regularized normalization (KL-Norm) which make the normalized data well behaved and helps in better generalization as it reduces over-fitting, generalises well on out of domain distributions and removes irrelevant biases and features with negligible increase in model parameters and memory overheads. Detailed experimental evaluation on multiple low resource NLP and speech tasks, demonstrates the superior performance of KL-Norm as compared to other popular normalization and regularization techniques.