 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeARIA: Adaptive Region-Based Importance Allocation for Conditional Diffusion Distillation
Jun 22, 2026Distilling conditional diffusion models aims to transfer the behavior of a large teacher to a smaller student while preserving alignment across conditioning inputs. Unlike recognition tasks, knowledge distillation in conditional diffusion often struggles to transfer knowledge beyond the training distribution, since the predicted noise strongly depends on the conditioning signal. As a result, effective distillation requires exploring a large conditioning space. In practical settings, this creates a major bottleneck. Paired image-condition data may be limited, and generating synthetic images for every available condition is often computationally infeasible, while the pool of conditions, such as text prompts, can be extremely large. Recent work addresses this issue by switching conditions during training, exposing the student to a broader conditioning space without changing the distillation objective. Yet this raises a complementary question: once a large conditioning corpus is available, how should the training effort be allocated? In this work, we introduce ARIA, a framework that adaptively allocates training effort across coarse regions of the conditioning space. By maintaining online estimates of teacher-student discrepancy at the region level, ARIA focuses updates where misalignment persists while preserving the original distillation objective. Empirically, ARIA improves over RC across most architectures and settings, with the clearest gains observed in unseen and underrepresented regimes. We also provide a theoretical analysis showing that the proposed tracking mechanism follows the evolving discrepancy during training under bounded variance and drift assumptions.
Self-Improving VLA Policies: Selected Diffusion Noise for Spurious-Robust Action Smoothing
Jun 12, 2026Diffusion-based Vision-Language-Action (VLA) policies enable strong generalization in robotic manipulation, but remain sensitive to spurious visual correlations and noisy action generation, leading to brittle behavior under perturbations. We introduce Selected Diffusion Noise (SDN), a simple, training-free test-time method that improves both robustness and success rate by leveraging the diffusion noise space as a controllable degree of freedom. SDN dynamically samples noise vectors that are maximally separated from a reference set to mitigate reliance on spurious cues, while selecting candidates that yield more coherent action trajectories. This dual objective encourages stable behavior even under object-masked observations and reduces action jitter without modifying model parameters. We evaluate SDN on two simulation benchmarks (Google Robot, Widow-X) and two real-world robotic datasets across multiple VLA policies, including pi_0, Groot-N1.5, and Groot-N1.6. SDN consistently improves success rates by +8% in simulation and +10% in real-world settings, while producing smoother and more stable actions. Our results highlight that diffusion noise selection can serve as an effective and general mechanism for enhancing VLA policies at test time.
Protein Fold Classification at Scale: Benchmarking and Pretraining
May 18, 2026Classifying protein topology is essential for deciphering biological function, but progress is held back by the lack of large-scale benchmarks that avoid duplicates and by models that do not scale well. We introduce TEDBench, a large-scale, non-redundant benchmark for protein fold classification constructed from the Encyclopedia of Domains (TED) and Foldseek-clustered AlphaFold structures. We show that on TEDBench, current protein representation learning methods either require very large models or fail to deliver strong performance. To address this challenge, we propose Masked Invariant Autoencoders (MiAE), a self-supervised framework for protein structure representation learning. MiAE uses an extremely high masking ratio of up to 90% with an $\mathrm{SE(3)}$-invariant encoder and a lightweight decoder that reconstructs backbone coordinates from the latent representation and mask tokens. MiAE scales well and outperforms supervised counterparts and state-of-the-art baselines on TEDBench, establishing a strong recipe for protein fold classification. To test transfer beyond AlphaFold structures, we further benchmark on a curated dataset from experimental structures of CATH v4.4. TEDBench is available at https://github.com/BorgwardtLab/TEDBench.
SymDrift: One-Shot Generative Modeling under Symmetries
May 07, 2026Generative modeling of physical systems, such as molecules, requires learning distributions that are invariant under global symmetries, such as rotations in three-dimensional space. Equivariant diffusion and flow matching models can incorporate such invariances effectively, even when trained on a non-invariant empirical distribution, but they typically rely on costly multi-step sampling. Recently, drifting models have emerged as an efficient alternative, enabling single-step generation and achieving state-of-the-art performance in generative modeling tasks. However, we show that drifting models face a symmetry-specific challenge, since an equivariant generator does not generally produce the same drifting field as the one obtained from the symmetrized target distribution. Addressing this issue would require expensive symmetrization of the empirical distribution. To avoid this cost, we propose SymDrift, a framework that makes the drifting field itself symmetry-aware. We introduce two complementary strategies: (i) a symmetrized drift in coordinate space based on optimal alignment, and (ii) a $G$-invariant embedding that removes symmetry ambiguity by construction. Empirically, SymDrift outperforms existing one-shot methods on standard benchmarks for conformer and transition state generation, while remaining competitive with significantly more expensive multi-step approaches. By enabling one-shot inference, SymDrift reduces computational overhead by up to 40$\times$ compared to existing baselines, making it promising for high-throughput applications such as virtual drug screening and large-scale reaction network exploration.
Logical Guidance for the Exact Composition of Diffusion Models
Feb 05, 2026We propose LOGDIFF (Logical Guidance for the Exact Composition of Diffusion Models), a guidance framework for diffusion models that enables principled constrained generation with complex logical expressions at inference time. We study when exact score-based guidance for complex logical formulas can be obtained from guidance signals associated with atomic properties. First, we derive an exact Boolean calculus that provides a sufficient condition for exact logical guidance. Specifically, if a formula admits a circuit representation in which conjunctions combine conditionally independent subformulas and disjunctions combine subformulas that are either conditionally independent or mutually exclusive, exact logical guidance is achievable. In this case, the guidance signal can be computed exactly from atomic scores and posterior probabilities using an efficient recursive algorithm. Moreover, we show that, for commonly encountered classes of distributions, any desired Boolean formula is compilable into such a circuit representation. Second, by combining atomic guidance scores with posterior probability estimates, we introduce a hybrid guidance approach that bridges classifierguidance and classifier-free guidance, applicable to both compositional logical guidance and standard conditional generation. We demonstrate the effectiveness of our framework on multiple image and protein structure generation tasks.
SMART: Scalable Mesh-free Aerodynamic Simulations from Raw Geometries using a Transformer-based Surrogate Model
Jan 26, 2026Machine learning-based surrogate models have emerged as more efficient alternatives to numerical solvers for physical simulations over complex geometries, such as car bodies. Many existing models incorporate the simulation mesh as an additional input, thereby reducing prediction errors. However, generating a simulation mesh for new geometries is computationally costly. In contrast, mesh-free methods, which do not rely on the simulation mesh, typically incur higher errors. Motivated by these considerations, we introduce SMART, a neural surrogate model that predicts physical quantities at arbitrary query locations using only a point-cloud representation of the geometry, without requiring access to the simulation mesh. The geometry and simulation parameters are encoded into a shared latent space that captures both structural and parametric characteristics of the physical field. A physics decoder then attends to the encoder's intermediate latent representations to map spatial queries to physical quantities. Through this cross-layer interaction, the model jointly updates latent geometric features and the evolving physical field. Extensive experiments show that SMART is competitive with and often outperforms existing methods that rely on the simulation mesh as input, demonstrating its capabilities for industry-level simulations.
GraIP: A Benchmarking Framework For Neural Graph Inverse Problems
Jan 26, 2026A wide range of graph learning tasks, such as structure discovery, temporal graph analysis, and combinatorial optimization, focus on inferring graph structures from data, rather than making predictions on given graphs. However, the respective methods to solve such problems are often developed in an isolated, task-specific manner and thus lack a unifying theoretical foundation. Here, we provide a stepping stone towards the formation of such a foundation and further development by introducing the Neural Graph Inverse Problem (GraIP) conceptual framework, which formalizes and reframes a broad class of graph learning tasks as inverse problems. Unlike discriminative approaches that directly predict target variables from given graph inputs, the GraIP paradigm addresses inverse problems, i.e., it relies on observational data and aims to recover the underlying graph structure by reversing the forward process, such as message passing or network dynamics, that produced the observed outputs. We demonstrate the versatility of GraIP across various graph learning tasks, including rewiring, causal discovery, and neural relational inference. We also propose benchmark datasets and metrics for each GraIP domain considered, and characterize and empirically evaluate existing baseline methods used to solve them. Overall, our unifying perspective bridges seemingly disparate applications and provides a principled approach to structural learning in constrained and combinatorial settings while encouraging cross-pollination of existing methods across graph inverse problems.
How Many Tokens Do 3D Point Cloud Transformer Architectures Really Need?
Nov 07, 2025Recent advances in 3D point cloud transformers have led to state-of-the-art results in tasks such as semantic segmentation and reconstruction. However, these models typically rely on dense token representations, incurring high computational and memory costs during training and inference. In this work, we present the finding that tokens are remarkably redundant, leading to substantial inefficiency. We introduce gitmerge3D, a globally informed graph token merging method that can reduce the token count by up to 90-95% while maintaining competitive performance. This finding challenges the prevailing assumption that more tokens inherently yield better performance and highlights that many current models are over-tokenized and under-optimized for scalability. We validate our method across multiple 3D vision tasks and show consistent improvements in computational efficiency. This work is the first to assess redundancy in large-scale 3D transformer models, providing insights into the development of more efficient 3D foundation architectures. Our code and checkpoints are publicly available at https://gitmerge3d.github.io
S-Chain: Structured Visual Chain-of-Thought For Medicine
Oct 26, 2025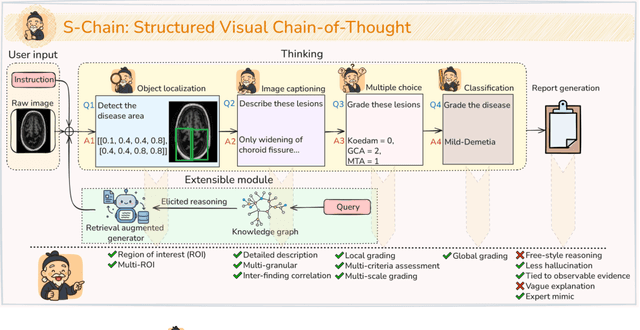
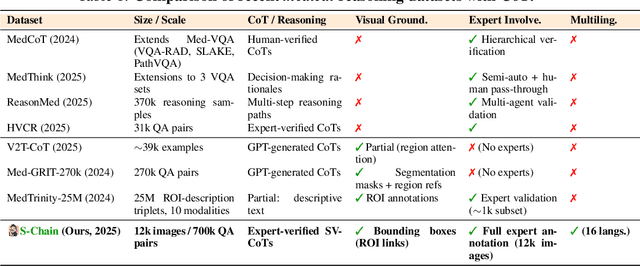
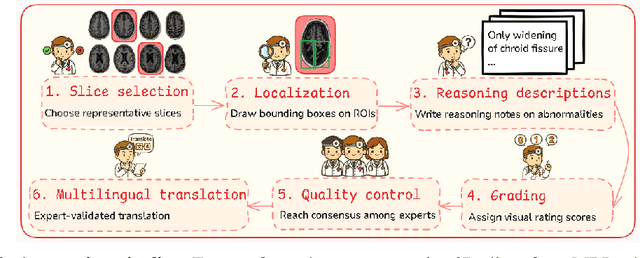
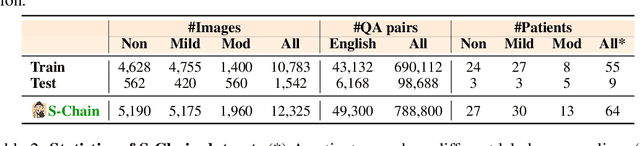
Faithful reasoning in medical vision-language models (VLMs) requires not only accurate predictions but also transparent alignment between textual rationales and visual evidence. While Chain-of-Thought (CoT) prompting has shown promise in medical visual question answering (VQA), no large-scale expert-level dataset has captured stepwise reasoning with precise visual grounding. We introduce S-Chain, the first large-scale dataset of 12,000 expert-annotated medical images with bounding boxes and structured visual CoT (SV-CoT), explicitly linking visual regions to reasoning steps. The dataset further supports 16 languages, totaling over 700k VQA pairs for broad multilingual applicability. Using S-Chain, we benchmark state-of-the-art medical VLMs (ExGra-Med, LLaVA-Med) and general-purpose VLMs (Qwen2.5-VL, InternVL2.5), showing that SV-CoT supervision significantly improves interpretability, grounding fidelity, and robustness. Beyond benchmarking, we study its synergy with retrieval-augmented generation, revealing how domain knowledge and visual grounding interact during autoregressive reasoning. Finally, we propose a new mechanism that strengthens the alignment between visual evidence and reasoning, improving both reliability and efficiency. S-Chain establishes a new benchmark for grounded medical reasoning and paves the way toward more trustworthy and explainable medical VLMs.
Learning the Neighborhood: Contrast-Free Multimodal Self-Supervised Molecular Graph Pretraining
Sep 26, 2025



High-quality molecular representations are essential for property prediction and molecular design, yet large labeled datasets remain scarce. While self-supervised pretraining on molecular graphs has shown promise, many existing approaches either depend on hand-crafted augmentations or complex generative objectives, and often rely solely on 2D topology, leaving valuable 3D structural information underutilized. To address this gap, we introduce C-FREE (Contrast-Free Representation learning on Ego-nets), a simple framework that integrates 2D graphs with ensembles of 3D conformers. C-FREE learns molecular representations by predicting subgraph embeddings from their complementary neighborhoods in the latent space, using fixed-radius ego-nets as modeling units across different conformers. This design allows us to integrate both geometric and topological information within a hybrid Graph Neural Network (GNN)-Transformer backbone, without negatives, positional encodings, or expensive pre-processing. Pretraining on the GEOM dataset, which provides rich 3D conformational diversity, C-FREE achieves state-of-the-art results on MoleculeNet, surpassing contrastive, generative, and other multimodal self-supervised methods. Fine-tuning across datasets with diverse sizes and molecule types further demonstrates that pretraining transfers effectively to new chemical domains, highlighting the importance of 3D-informed molecular representations.