 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeLED down the rabbit hole: exploring the potential of global attention for biomedical multi-document summarisation
Sep 19, 2022
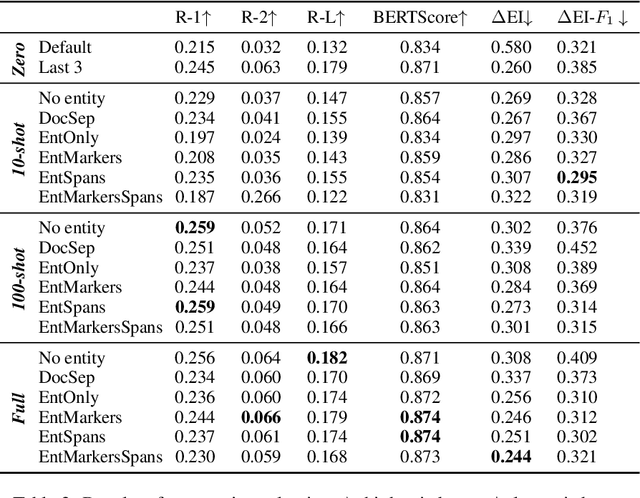
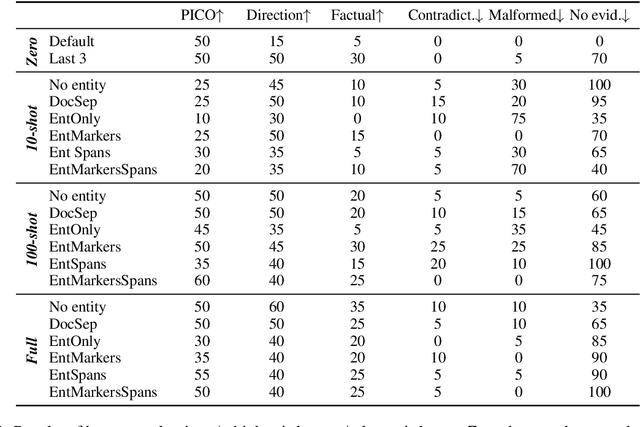
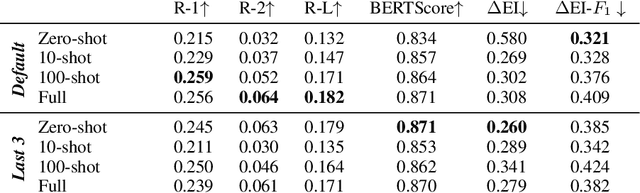
In this paper we report on our submission to the Multidocument Summarisation for Literature Review (MSLR) shared task. Specifically, we adapt PRIMERA (Xiao et al., 2022) to the biomedical domain by placing global attention on important biomedical entities in several ways. We analyse the outputs of the 23 resulting models, and report patterns in the results related to the presence of additional global attention, number of training steps, and the input configuration.
Cross-modal Clinical Graph Transformer for Ophthalmic Report Generation
Jun 04, 2022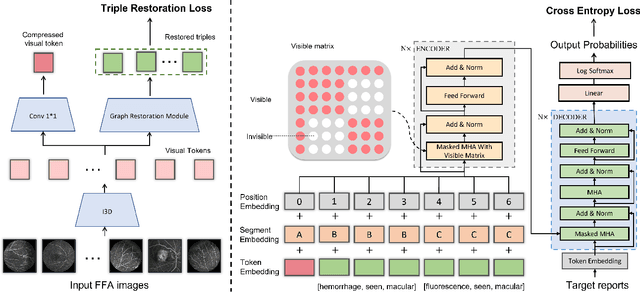

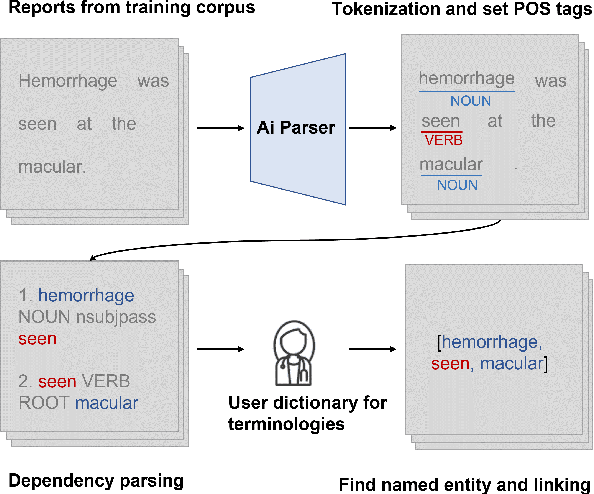
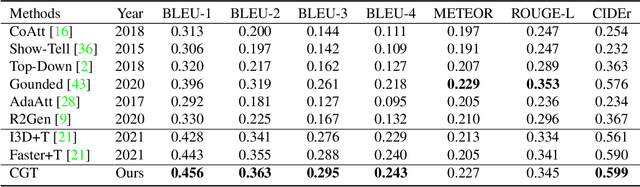
Automatic generation of ophthalmic reports using data-driven neural networks has great potential in clinical practice. When writing a report, ophthalmologists make inferences with prior clinical knowledge. This knowledge has been neglected in prior medical report generation methods. To endow models with the capability of incorporating expert knowledge, we propose a Cross-modal clinical Graph Transformer (CGT) for ophthalmic report generation (ORG), in which clinical relation triples are injected into the visual features as prior knowledge to drive the decoding procedure. However, two major common Knowledge Noise (KN) issues may affect models' effectiveness. 1) Existing general biomedical knowledge bases such as the UMLS may not align meaningfully to the specific context and language of the report, limiting their utility for knowledge injection. 2) Incorporating too much knowledge may divert the visual features from their correct meaning. To overcome these limitations, we design an automatic information extraction scheme based on natural language processing to obtain clinical entities and relations directly from in-domain training reports. Given a set of ophthalmic images, our CGT first restores a sub-graph from the clinical graph and injects the restored triples into visual features. Then visible matrix is employed during the encoding procedure to limit the impact of knowledge. Finally, reports are predicted by the encoded cross-modal features via a Transformer decoder. Extensive experiments on the large-scale FFA-IR benchmark demonstrate that the proposed CGT is able to outperform previous benchmark methods and achieve state-of-the-art performances.
Improving negation detection with negation-focused pre-training
May 09, 2022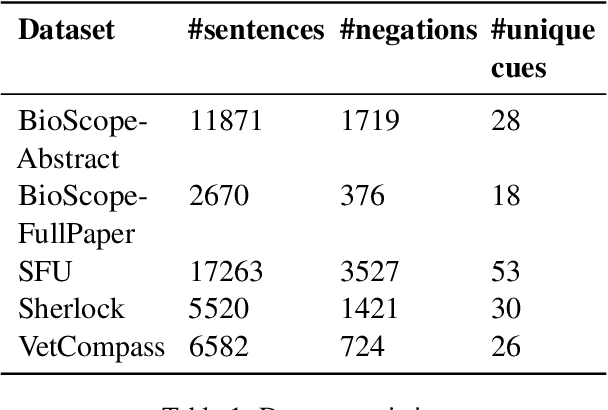
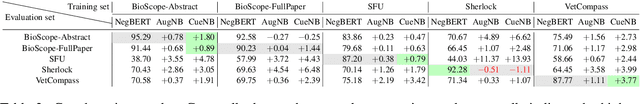
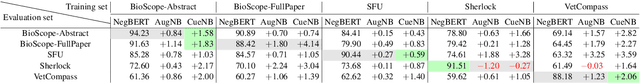
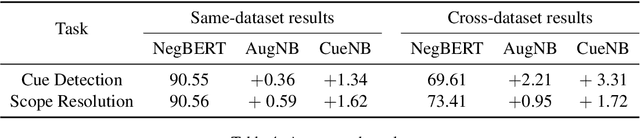
Negation is a common linguistic feature that is crucial in many language understanding tasks, yet it remains a hard problem due to diversity in its expression in different types of text. Recent work has shown that state-of-the-art NLP models underperform on samples containing negation in various tasks, and that negation detection models do not transfer well across domains. We propose a new negation-focused pre-training strategy, involving targeted data augmentation and negation masking, to better incorporate negation information into language models. Extensive experiments on common benchmarks show that our proposed approach improves negation detection performance and generalizability over the strong baseline NegBERT (Khandewal and Sawant, 2020).
ITTC @ TREC 2021 Clinical Trials Track
Feb 16, 2022
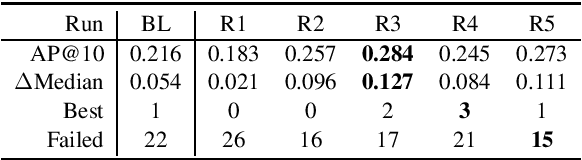
This paper describes the submissions of the Natural Language Processing (NLP) team from the Australian Research Council Industrial Transformation Training Centre (ITTC) for Cognitive Computing in Medical Technologies to the TREC 2021 Clinical Trials Track. The task focuses on the problem of matching eligible clinical trials to topics constituting a summary of a patient's admission notes. We explore different ways of representing trials and topics using NLP techniques, and then use a common retrieval model to generate the ranked list of relevant trials for each topic. The results from all our submitted runs are well above the median scores for all topics, but there is still plenty of scope for improvement.
MPVNN: Mutated Pathway Visible Neural Network Architecture for Interpretable Prediction of Cancer-specific Survival Risk
Feb 02, 2022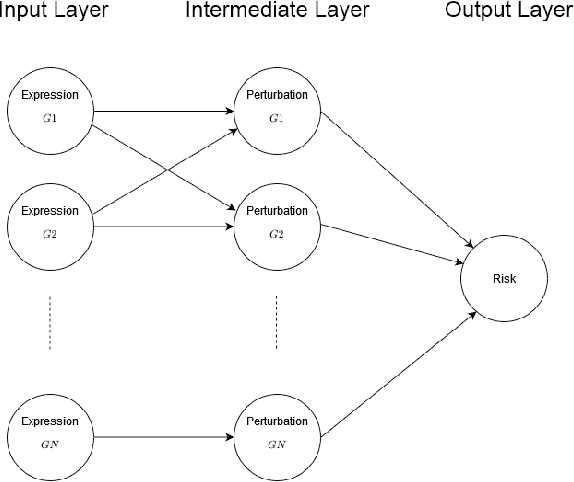
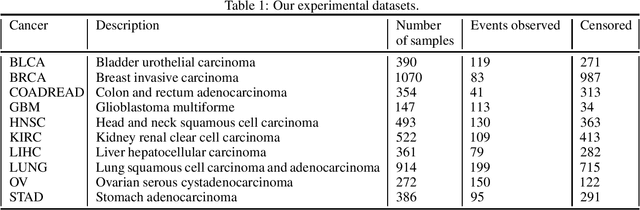
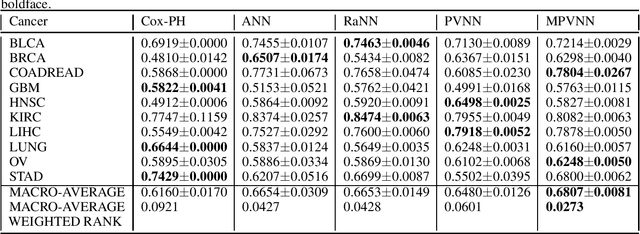
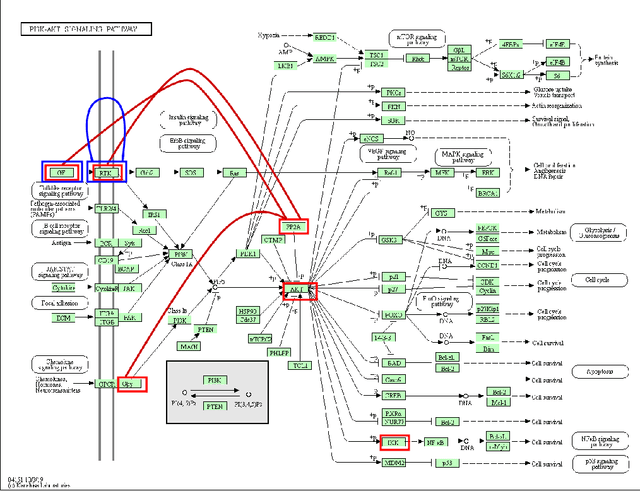
Survival risk prediction using gene expression data is important in making treatment decisions in cancer. Standard neural network (NN) survival analysis models are black boxes with lack of interpretability. More interpretable visible neural network (VNN) architectures are designed using biological pathway knowledge. But they do not model how pathway structures can change for particular cancer types. We propose a novel Mutated Pathway VNN or MPVNN architecture, designed using prior signaling pathway knowledge and gene mutation data-based edge randomization simulating signal flow disruption. As a case study, we use the PI3K-Akt pathway and demonstrate overall improved cancer-specific survival risk prediction results of MPVNN over standard non-NN and other similar sized NN survival analysis methods. We show that trained MPVNN architecture interpretation, which points to smaller sets of genes connected by signal flow within the PI3K-Akt pathway that are important in risk prediction for particular cancer types, is reliable.
Large-scale protein-protein post-translational modification extraction with distant supervision and confidence calibrated BioBERT
Jan 06, 2022
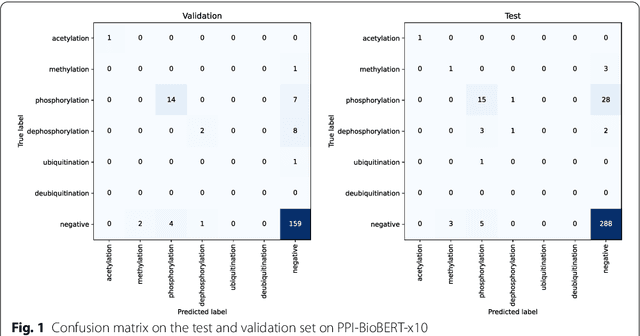
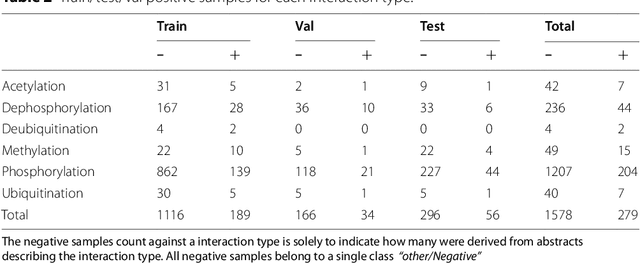
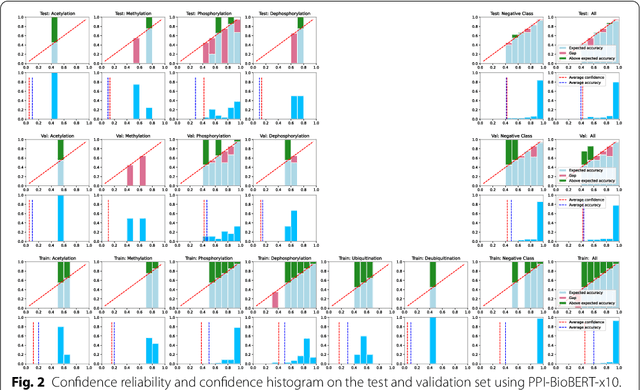
Protein-protein interactions (PPIs) are critical to normal cellular function and are related to many disease pathways. However, only 4% of PPIs are annotated with PTMs in biological knowledge databases such as IntAct, mainly performed through manual curation, which is neither time nor cost-effective. We use the IntAct PPI database to create a distant supervised dataset annotated with interacting protein pairs, their corresponding PTM type, and associated abstracts from the PubMed database. We train an ensemble of BioBERT models - dubbed PPI-BioBERT-x10 to improve confidence calibration. We extend the use of ensemble average confidence approach with confidence variation to counteract the effects of class imbalance to extract high confidence predictions. The PPI-BioBERT-x10 model evaluated on the test set resulted in a modest F1-micro 41.3 (P =5 8.1, R = 32.1). However, by combining high confidence and low variation to identify high quality predictions, tuning the predictions for precision, we retained 19% of the test predictions with 100% precision. We evaluated PPI-BioBERT-x10 on 18 million PubMed abstracts and extracted 1.6 million (546507 unique PTM-PPI triplets) PTM-PPI predictions, and filter ~ 5700 (4584 unique) high confidence predictions. Of the 5700, human evaluation on a small randomly sampled subset shows that the precision drops to 33.7% despite confidence calibration and highlights the challenges of generalisability beyond the test set even with confidence calibration. We circumvent the problem by only including predictions associated with multiple papers, improving the precision to 58.8%. In this work, we highlight the benefits and challenges of deep learning-based text mining in practice, and the need for increased emphasis on confidence calibration to facilitate human curation efforts.
* BMC BioInformatics
Impact of detecting clinical trial elements in exploration of COVID-19 literature
May 25, 2021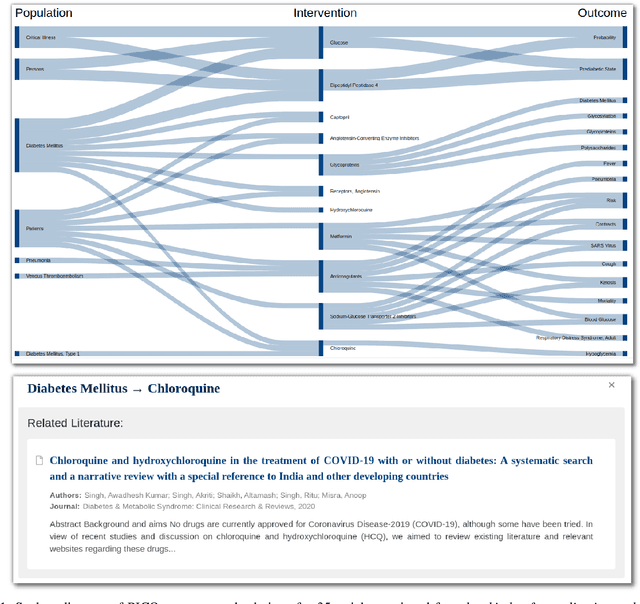
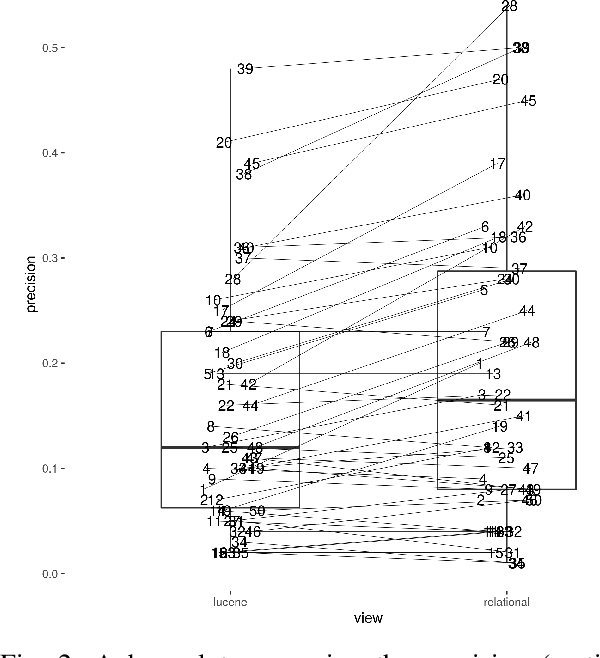
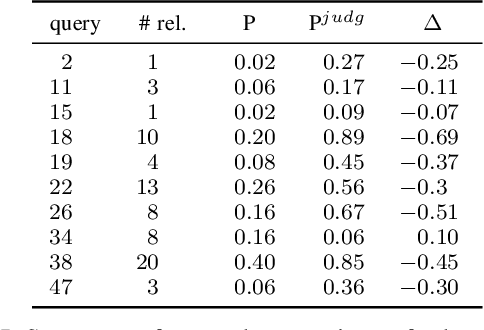

The COVID-19 pandemic has driven ever-greater demand for tools which enable efficient exploration of biomedical literature. Although semi-structured information resulting from concept recognition and detection of the defining elements of clinical trials (e.g. PICO criteria) has been commonly used to support literature search, the contributions of this abstraction remain poorly understood, especially in relation to text-based retrieval. In this study, we compare the results retrieved by a standard search engine with those filtered using clinically-relevant concepts and their relations. With analysis based on the annotations from the TREC-COVID shared task, we obtain quantitative as well as qualitative insights into characteristics of relational and concept-based literature exploration. Most importantly, we find that the relational concept selection filters the original retrieved collection in a way that decreases the proportion of unjudged documents and increases the precision, which means that the user is likely to be exposed to a larger number of relevant documents.
Memorization vs. Generalization: Quantifying Data Leakage in NLP Performance Evaluation
Feb 03, 2021
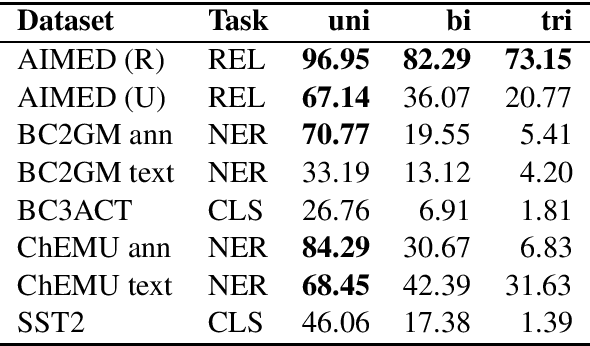
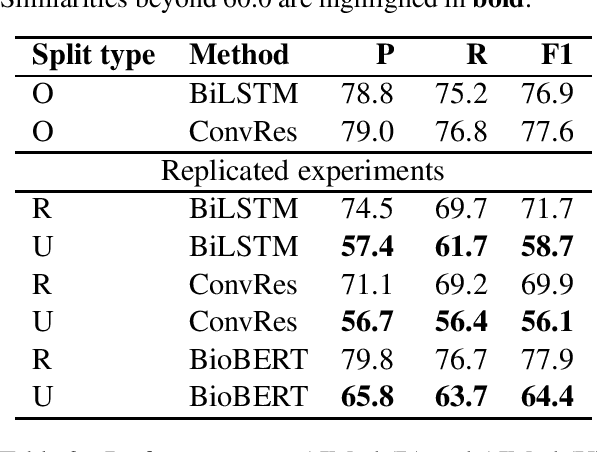
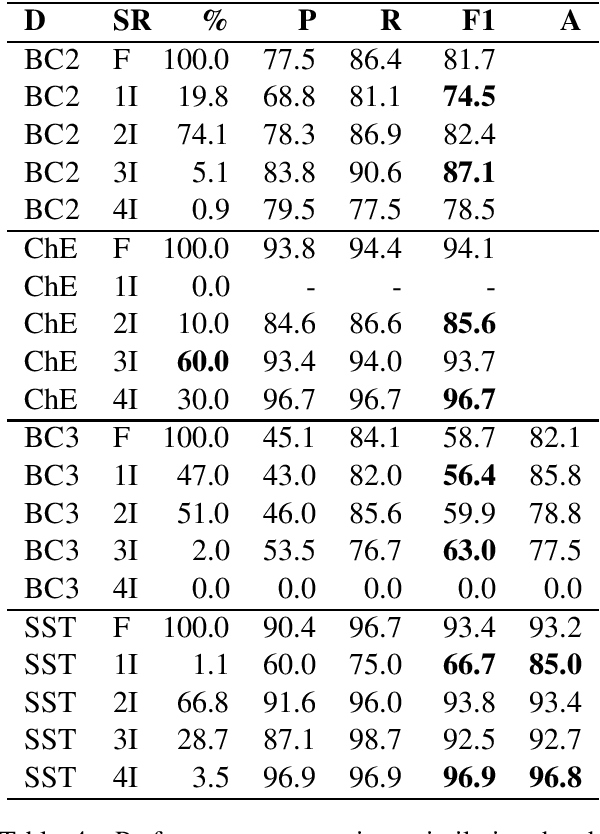
Public datasets are often used to evaluate the efficacy and generalizability of state-of-the-art methods for many tasks in natural language processing (NLP). However, the presence of overlap between the train and test datasets can lead to inflated results, inadvertently evaluating the model's ability to memorize and interpreting it as the ability to generalize. In addition, such data sets may not provide an effective indicator of the performance of these methods in real world scenarios. We identify leakage of training data into test data on several publicly available datasets used to evaluate NLP tasks, including named entity recognition and relation extraction, and study them to assess the impact of that leakage on the model's ability to memorize versus generalize.
Assigning function to protein-protein interactions: a weakly supervised BioBERT based approach using PubMed abstracts
Sep 29, 2020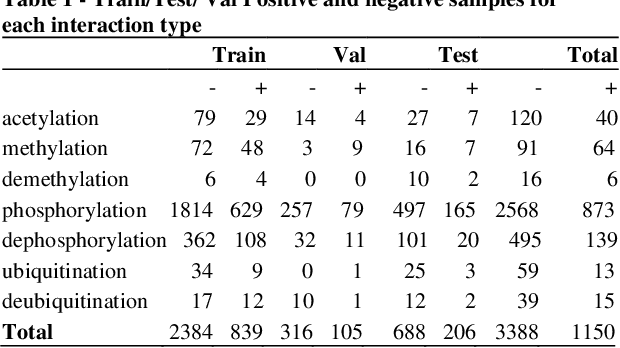
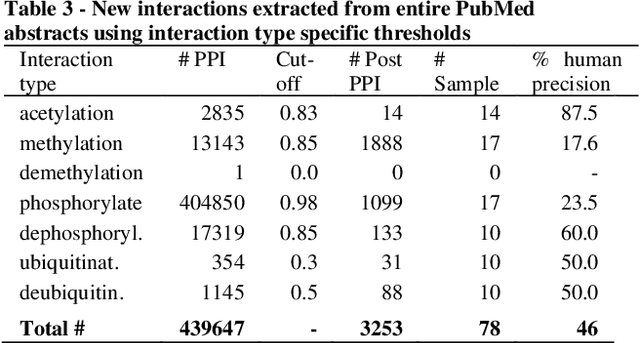
Motivation: Protein-protein interactions (PPI) are critical to the function of proteins in both normal and diseased cells, and many critical protein functions are mediated by interactions.Knowledge of the nature of these interactions is important for the construction of networks to analyse biological data. However, only a small percentage of PPIs captured in protein interaction databases have annotations of function available, e.g. only 4% of PPI are functionally annotated in the IntAct database. Here, we aim to label the function type of PPIs by extracting relationships described in PubMed abstracts. Method: We create a weakly supervised dataset from the IntAct PPI database containing interacting protein pairs with annotated function and associated abstracts from the PubMed database. We apply a state-of-the-art deep learning technique for biomedical natural language processing tasks, BioBERT, to build a model - dubbed PPI-BioBERT - for identifying the function of PPIs. In order to extract high quality PPI functions at large scale, we use an ensemble of PPI-BioBERT models to improve uncertainty estimation and apply an interaction type-specific threshold to counteract the effects of variations in the number of training samples per interaction type. Results: We scan 18 million PubMed abstracts to automatically identify 3253 new typed PPIs, including phosphorylation and acetylation interactions, with an overall precision of 46% (87% for acetylation) based on a human-reviewed sample. This work demonstrates that analysis of biomedical abstracts for PPI function extraction is a feasible approach to substantially increasing the number of interactions annotated with function captured in online databases.
COVID-SEE: Scientific Evidence Explorer for COVID-19 Related Research
Aug 18, 2020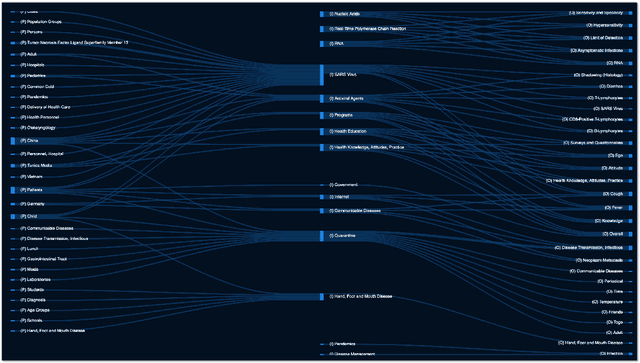
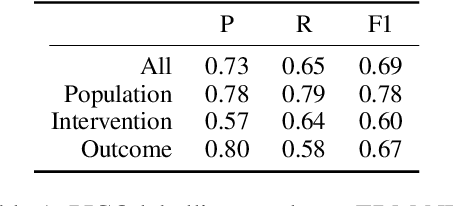

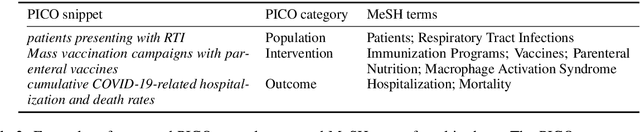
We present COVID-SEE, a system for medical literature discovery based on the concept of information exploration, which builds on several distinct text analysis and natural language processing methods to structure and organise information in publications, and augments search by providing a visual overview supporting exploration of a collection to identify key articles of interest. We developed this system over COVID-19 literature to help medical professionals and researchers explore the literature evidence, and improve findability of relevant information. COVID-SEE is available at http://covid-see.com.