 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGraph Information Bottleneck
Oct 24, 2020
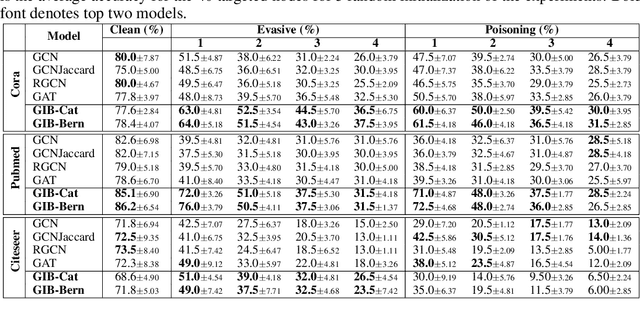
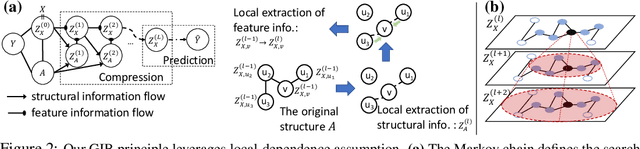
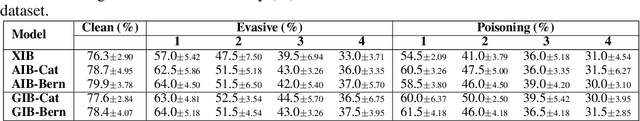
Representation learning of graph-structured data is challenging because both graph structure and node features carry important information. Graph Neural Networks (GNNs) provide an expressive way to fuse information from network structure and node features. However, GNNs are prone to adversarial attacks. Here we introduce Graph Information Bottleneck (GIB), an information-theoretic principle that optimally balances expressiveness and robustness of the learned representation of graph-structured data. Inheriting from the general Information Bottleneck (IB), GIB aims to learn the minimal sufficient representation for a given task by maximizing the mutual information between the representation and the target, and simultaneously constraining the mutual information between the representation and the input data. Different from the general IB, GIB regularizes the structural as well as the feature information. We design two sampling algorithms for structural regularization and instantiate the GIB principle with two new models: GIB-Cat and GIB-Bern, and demonstrate the benefits by evaluating the resilience to adversarial attacks. We show that our proposed models are more robust than state-of-the-art graph defense models. GIB-based models empirically achieve up to 31% improvement with adversarial perturbation of the graph structure as well as node features.
Beta Embeddings for Multi-Hop Logical Reasoning in Knowledge Graphs
Oct 22, 2020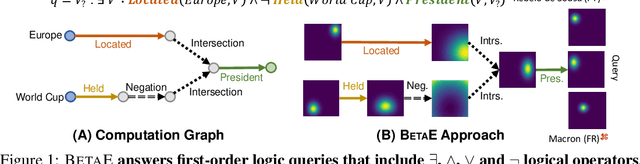
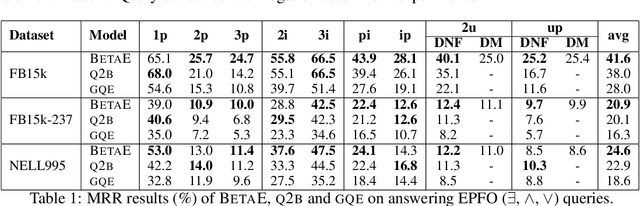
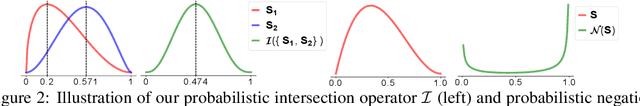
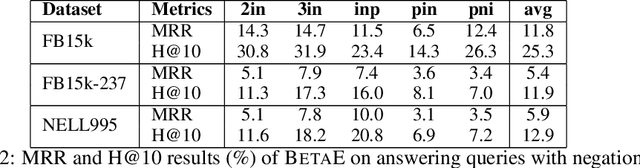
One of the fundamental problems in Artificial Intelligence is to perform complex multi-hop logical reasoning over the facts captured by a knowledge graph (KG). This problem is challenging, because KGs can be massive and incomplete. Recent approaches embed KG entities in a low dimensional space and then use these embeddings to find the answer entities. However, it has been an outstanding challenge of how to handle arbitrary first-order logic (FOL) queries as present methods are limited to only a subset of FOL operators. In particular, the negation operator is not supported. An additional limitation of present methods is also that they cannot naturally model uncertainty. Here, we present BetaE, a probabilistic embedding framework for answering arbitrary FOL queries over KGs. BetaE is the first method that can handle a complete set of first-order logical operations: conjunction ($\wedge$), disjunction ($\vee$), and negation ($\neg$). A key insight of BetaE is to use probabilistic distributions with bounded support, specifically the Beta distribution, and embed queries/entities as distributions, which as a consequence allows us to also faithfully model uncertainty. Logical operations are performed in the embedding space by neural operators over the probabilistic embeddings. We demonstrate the performance of BetaE on answering arbitrary FOL queries on three large, incomplete KGs. While being more general, BetaE also increases relative performance by up to 25.4% over the current state-of-the-art KG reasoning methods that can only handle conjunctive queries without negation.
Direct Multi-hop Attention based Graph Neural Network
Oct 02, 2020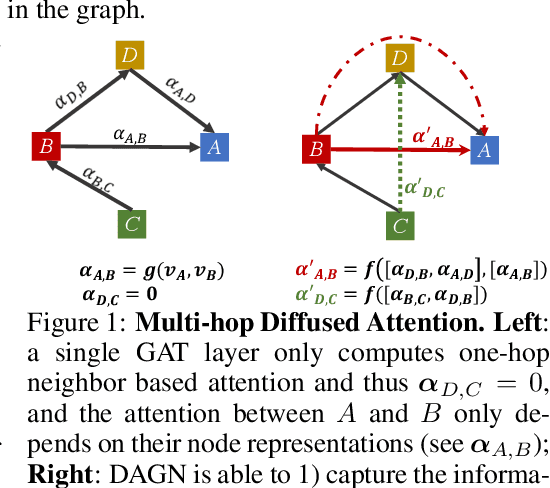
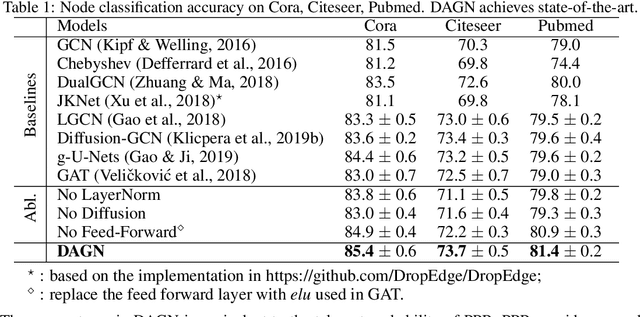
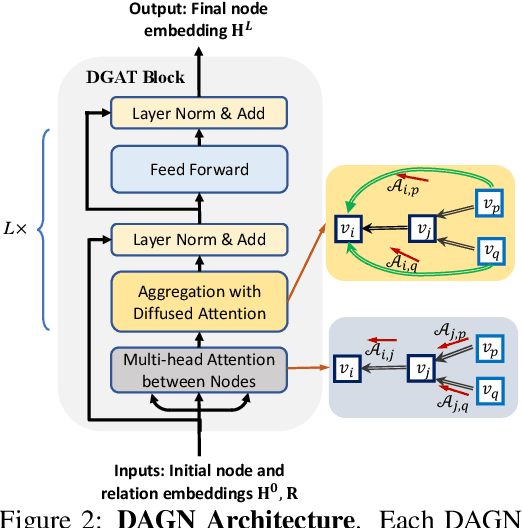
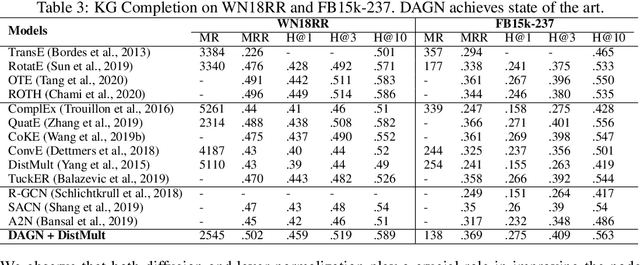
Introducing self-attention mechanism in graph neural networks (GNNs) achieved state-of-the-art performance for graph representation learning. However, at every layer, attention is only computed between two connected nodes and depends solely on the representation of both nodes. This attention computation cannot account for the multi-hop neighbors which supply graph structure context information and have influence on the node representation learning as well. In this paper, we propose Direct Multi-hop Attention based Graph neural Network (DAGN) for graph representation learning, a principled way to incorporate multi-hop neighboring context into attention computation, enabling long-range interactions at every layer. To compute attention between nodes that are multiple hops away, DAGN diffuses the attention scores from neighboring nodes to non-neighboring nodes, thus increasing the receptive field for every message passing layer. Unlike previous methods, DAGN uses a diffusion prior on attention values, to efficiently account for all paths between the pair of nodes when computing multi-hop attention weights. This helps DAGN capture large-scale structural information in a single layer, and learn more informative attention distribution. Experimental results on standard semi-supervised node classification as well as the knowledge graph completion show that DAGN achieves state-of-the-art results: DAGN achieves up to 5.7% relative error reduction over the previous state-of-the-art on Cora, Citeseer, and Pubmed. DAGN also obtains the best performance on a large-scale Open Graph Benchmark dataset. On knowledge graph completion DAGN advances state-of-the-art on WN18RR and FB15k-237 across four different performance metrics.
Inductive Learning on Commonsense Knowledge Graph Completion
Sep 19, 2020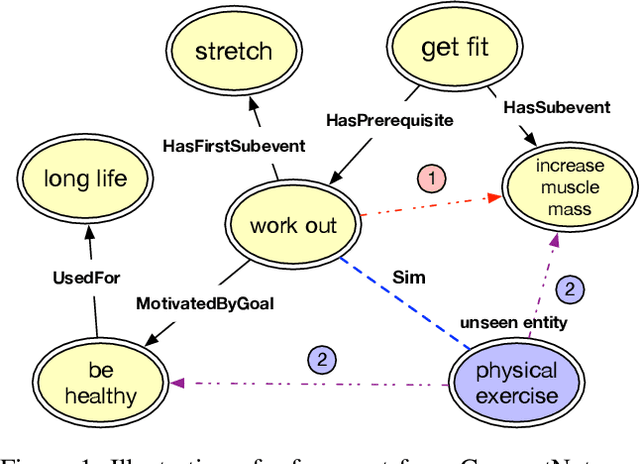
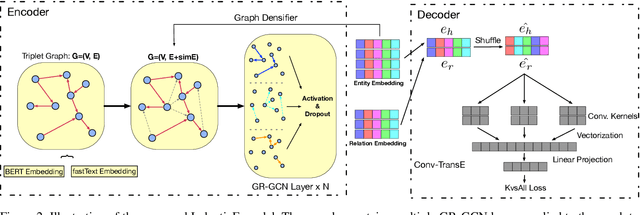
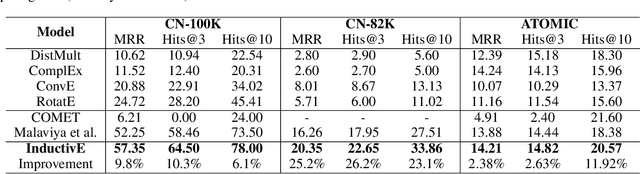
Commonsense knowledge graph (CKG) is a special type of knowledge graph (KG), where entities are composed of free-form text. However, most existing CKG completion methods focus on the setting where all the entities are presented at training time. Although this setting is standard for conventional KG completion, it has limitations for CKG completion. At test time, entities in CKGs can be unseen because they may have unseen text/names and entities may be disconnected from the training graph, since CKGs are generally very sparse. Here, we propose to study the inductive learning setting for CKG completion where unseen entities may present at test time. We develop a novel learning framework named InductivE. Different from previous approaches, InductiveE ensures the inductive learning capability by directly computing entity embeddings from raw entity attributes/text. InductiveE consists of a free-text encoder, a graph encoder, and a KG completion decoder. Specifically, the free-text encoder first extracts the textual representation of each entity based on the pre-trained language model and word embedding. The graph encoder is a gated relational graph convolutional neural network that learns from a densified graph for more informative entity representation learning. We develop a method that densifies CKGs by adding edges among semantic-related entities and provide more supportive information for unseen entities, leading to better generalization ability of entity embedding for unseen entities. Finally, inductiveE employs Conv-TransE as the CKG completion decoder. Experimental results show that InductiveE significantly outperforms state-of-the-art baselines in both standard and inductive settings on ATOMIC and ConceptNet benchmarks. InductivE performs especially well on inductive scenarios where it achieves above 48% improvement over present methods.
Distance Encoding -- Design Provably More Powerful Graph Neural Networks for Structural Representation Learning
Sep 05, 2020
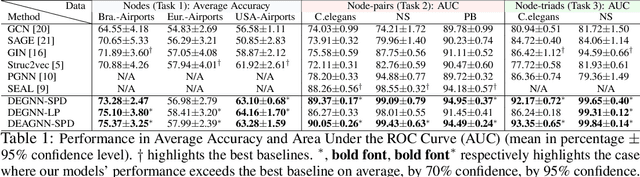
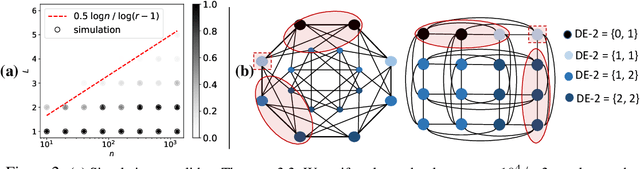
Learning structural representations of node sets from graph-structured data is crucial for applications ranging from node-role discovery to link prediction and molecule classification. Graph Neural Networks (GNNs) have achieved great success in structural representation learning. However, most GNNs are limited by the 1-Weisfeiler-Lehman (WL) test and thus possible to generate identical representation for structures and graphs that are actually different. More powerful GNNs, proposed recently by mimicking higher-order-WL tests, only focus on entire-graph representations and cannot utilize sparsity of the graph structure to be computationally efficient. Here we propose a general class of structure-related features, termed Distance Encoding (DE), to assist GNNs in representing node sets with arbitrary sizes with strictly more expressive power than the 1-WL test. DE essentially captures the distance between the node set whose representation is to be learnt and each node in the graph, which includes important graph-related measures such as shortest-path-distance and generalized PageRank scores. We propose two general frameworks for GNNs to use DEs (1) as extra node attributes and (2) further as controllers of message aggregation in GNNs. Both frameworks may still utilize the sparse structure to keep scalability to process large graphs. In theory, we prove that these two frameworks can distinguish node sets embedded in almost all regular graphs where traditional GNNs always fail. We also rigorously analyze their limitations. Empirically, we evaluate these two frameworks on node structural roles prediction, link prediction and triangle prediction over six real networks. The results show that our models outperform GNNs without DEs by up-to 15% improvement in average accuracy and AUC. Our models also significantly outperform other SOTA baselines particularly designed for those tasks.
OCEAN: Online Task Inference for Compositional Tasks with Context Adaptation
Aug 17, 2020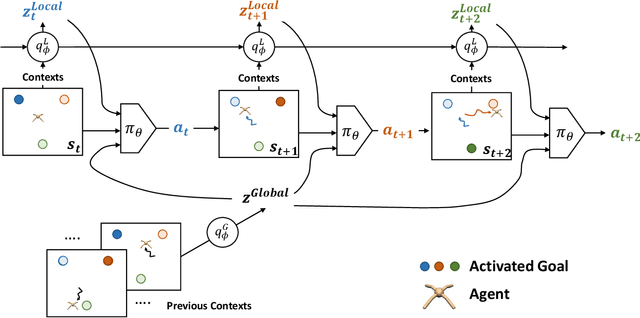
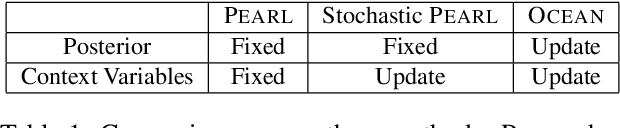

Real-world tasks often exhibit a compositional structure that contains a sequence of simpler sub-tasks. For instance, opening a door requires reaching, grasping, rotating, and pulling the door knob. Such compositional tasks require an agent to reason about the sub-task at hand while orchestrating global behavior accordingly. This can be cast as an online task inference problem, where the current task identity, represented by a context variable, is estimated from the agent's past experiences with probabilistic inference. Previous approaches have employed simple latent distributions, e.g., Gaussian, to model a single context for the entire task. However, this formulation lacks the expressiveness to capture the composition and transition of the sub-tasks. We propose a variational inference framework OCEAN to perform online task inference for compositional tasks. OCEAN models global and local context variables in a joint latent space, where the global variables represent a mixture of sub-tasks required for the task, while the local variables capture the transitions between the sub-tasks. Our framework supports flexible latent distributions based on prior knowledge of the task structure and can be trained in an unsupervised manner. Experimental results show that OCEAN provides more effective task inference with sequential context adaptation and thus leads to a performance boost on complex, multi-stage tasks.
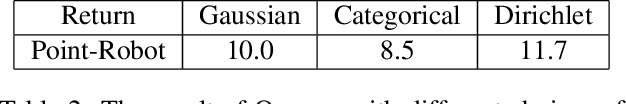
Concept Learners for Generalizable Few-Shot Learning
Jul 14, 2020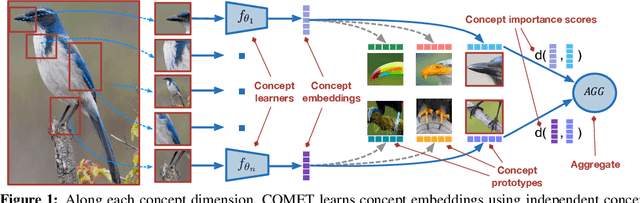
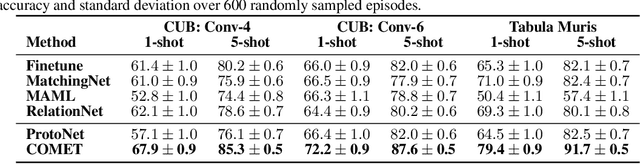

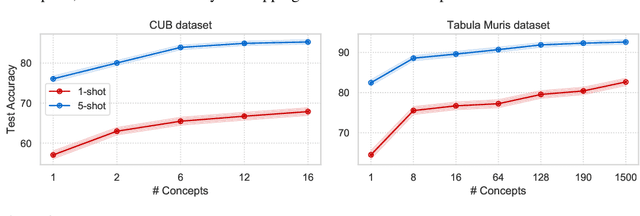
Developing algorithms that are able to generalize to a novel task given only a few labeled examples represents a fundamental challenge in closing the gap between machine- and human-level performance. The core of human cognition lies in the structured, reusable concepts that help us to rapidly adapt to new tasks and provide reasoning behind our decisions. However, existing meta-learning methods learn complex representations across prior labeled tasks without imposing any structure on the learned representations. Here we propose COMET, a meta-learning method that improves generalization ability by learning to learn along human-interpretable concept dimensions. Instead of learning a joint unstructured metric space, COMET learns mappings of high-level concepts into semi-structured metric spaces, and effectively combines the outputs of independent concept learners. We evaluate our model on few-shot tasks from diverse domains, including a benchmark image classification dataset and a novel single-cell dataset from a biological domain developed in our work. COMET significantly outperforms strong meta-learning baselines, achieving $9$-$12\%$ average improvement on the most challenging $1$-shot learning tasks, while unlike existing methods also providing interpretations behind the model's predictions.
Graph Structure of Neural Networks
Jul 13, 2020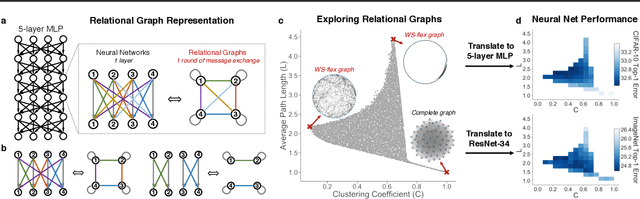

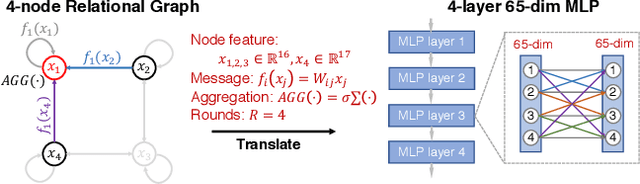
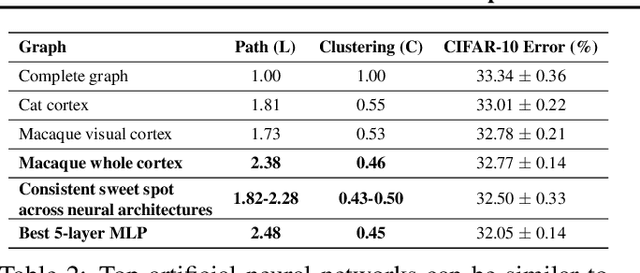
Neural networks are often represented as graphs of connections between neurons. However, despite their wide use, there is currently little understanding of the relationship between the graph structure of the neural network and its predictive performance. Here we systematically investigate how does the graph structure of neural networks affect their predictive performance. To this end, we develop a novel graph-based representation of neural networks called relational graph, where layers of neural network computation correspond to rounds of message exchange along the graph structure. Using this representation we show that: (1) a "sweet spot" of relational graphs leads to neural networks with significantly improved predictive performance; (2) neural network's performance is approximately a smooth function of the clustering coefficient and average path length of its relational graph; (3) our findings are consistent across many different tasks and datasets; (4) the sweet spot can be identified efficiently; (5) top-performing neural networks have graph structure surprisingly similar to those of real biological neural networks. Our work opens new directions for the design of neural architectures and the understanding on neural networks in general.
PinnerSage: Multi-Modal User Embedding Framework for Recommendations at Pinterest
Jul 07, 2020


Latent user representations are widely adopted in the tech industry for powering personalized recommender systems. Most prior work infers a single high dimensional embedding to represent a user, which is a good starting point but falls short in delivering a full understanding of the user's interests. In this work, we introduce PinnerSage, an end-to-end recommender system that represents each user via multi-modal embeddings and leverages this rich representation of users to provides high quality personalized recommendations. PinnerSage achieves this by clustering users' actions into conceptually coherent clusters with the help of a hierarchical clustering method (Ward) and summarizes the clusters via representative pins (Medoids) for efficiency and interpretability. PinnerSage is deployed in production at Pinterest and we outline the several design decisions that makes it run seamlessly at a very large scale. We conduct several offline and online A/B experiments to show that our method significantly outperforms single embedding methods.
* 10 pages, 7 figures
Neural Subgraph Matching
Jul 06, 2020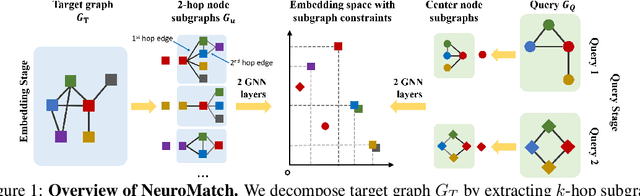
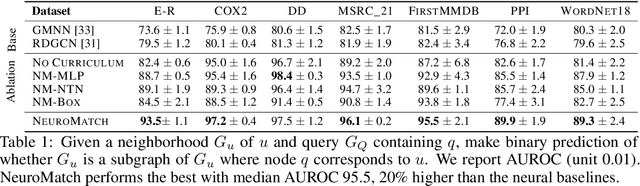

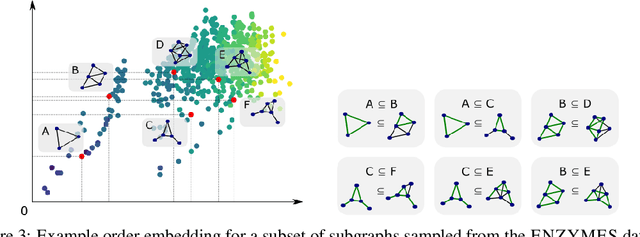
Subgraph matching is the problem of determining the presence and location(s) of a given query graph in a large target graph. Despite being an NP-complete problem, the subgraph matching problem is crucial in domains ranging from network science and database systems to biochemistry and cognitive science. However, existing techniques based on combinatorial matching and integer programming cannot handle matching problems with both large target and query graphs. Here we propose NeuroMatch, an accurate, efficient, and robust neural approach to subgraph matching. NeuroMatch decomposes query and target graphs into small subgraphs and embeds them using graph neural networks. Trained to capture geometric constraints corresponding to subgraph relations, NeuroMatch then efficiently performs subgraph matching directly in the embedding space. Experiments demonstrate NeuroMatch is 100x faster than existing combinatorial approaches and 18% more accurate than existing approximate subgraph matching methods.