 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeLeadsee-Precip: A Deep Learning Diagnostic Model for Precipitation
Nov 19, 2024



Recently, deep-learning weather forecasting models have surpassed traditional numerical models in terms of the accuracy of meteorological variables. However, there is considerable potential for improvements in precipitation forecasts, especially for heavy precipitation events. To address this deficiency, we propose Leadsee-Precip, a global deep learning model to generate precipitation from meteorological circulation fields. The model utilizes an information balance scheme to tackle the challenges of predicting heavy precipitation caused by the long-tail distribution of precipitation data. Additionally, more accurate satellite and radar-based precipitation retrievals are used as training targets. Compared to artificial intelligence global weather models, the heavy precipitation from Leadsee-Precip is more consistent with observations and shows competitive performance against global numerical weather prediction models. Leadsee-Precip can be integrated with any global circulation model to generate precipitation forecasts. But the deviations between the predicted and the ground-truth circulation fields may lead to a weakened precipitation forecast, which could potentially be mitigated by further fine-tuning based on the predicted circulation fields.
Improved AutoEncoder with LSTM module and KL divergence
Apr 30, 2024



The task of anomaly detection is to separate anomalous data from normal data in the dataset. Models such as deep convolutional autoencoder (CAE) network and deep supporting vector data description (SVDD) model have been universally employed and have demonstrated significant success in detecting anomalies. However, the over-reconstruction ability of CAE network for anomalous data can easily lead to high false negative rate in detecting anomalous data. On the other hand, the deep SVDD model has the drawback of feature collapse, which leads to a decrease of detection accuracy for anomalies. To address these problems, we propose the Improved AutoEncoder with LSTM module and Kullback-Leibler divergence (IAE-LSTM-KL) model in this paper. An LSTM network is added after the encoder to memorize feature representations of normal data. In the meanwhile, the phenomenon of feature collapse can also be mitigated by penalizing the featured input to SVDD module via KL divergence. The efficacy of the IAE-LSTM-KL model is validated through experiments on both synthetic and real-world datasets. Experimental results show that IAE-LSTM-KL model yields higher detection accuracy for anomalies. In addition, it is also found that the IAE-LSTM-KL model demonstrates enhanced robustness to contaminated outliers in the dataset.
Harmonic enhancement using learnable comb filter for light-weight full-band speech enhancement model
Jun 01, 2023



With fewer feature dimensions, filter banks are often used in light-weight full-band speech enhancement models. In order to further enhance the coarse speech in the sub-band domain, it is necessary to apply a post-filtering for harmonic retrieval. The signal processing-based comb filters used in RNNoise and PercepNet have limited performance and may cause speech quality degradation due to inaccurate fundamental frequency estimation. To tackle this problem, we propose a learnable comb filter to enhance harmonics. Based on the sub-band model, we design a DNN-based fundamental frequency estimator to estimate the discrete fundamental frequencies and a comb filter for harmonic enhancement, which are trained via an end-to-end pattern. The experiments show the advantages of our proposed method over PecepNet and DeepFilterNet.
Are Learned Molecular Representations Ready For Prime Time?
Apr 02, 2019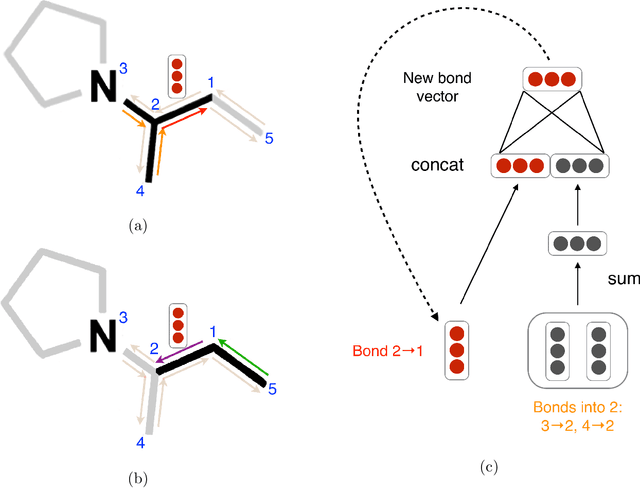


Advancements in neural machinery have led to a wide range of algorithmic solutions for molecular property prediction. Two classes of models in particular have yielded promising results: neural networks applied to computed molecular fingerprints or expert-crafted descriptors, and graph convolutional neural networks that construct a learned molecular representation by operating on the graph structure of the molecule. However, recent literature has yet to clearly determine which of these two methods is superior when generalizing to new chemical space. Furthermore, prior research has rarely examined these new models in industry research settings in comparison to existing employed models. In this paper, we benchmark models extensively on 19 public and 15 proprietary industrial datasets spanning a wide variety of chemical endpoints. In addition, we introduce a graph convolutional model that consistently outperforms models using fixed molecular descriptors as well as previous graph neural architectures on both public and proprietary datasets. Our empirical findings indicate that while approaches based on these representations have yet to reach the level of experimental reproducibility, our proposed model nevertheless offers significant improvements over models currently used in industrial workflows.
Chemi-net: a graph convolutional network for accurate drug property prediction
Mar 21, 2018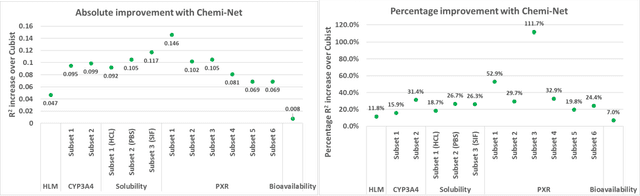
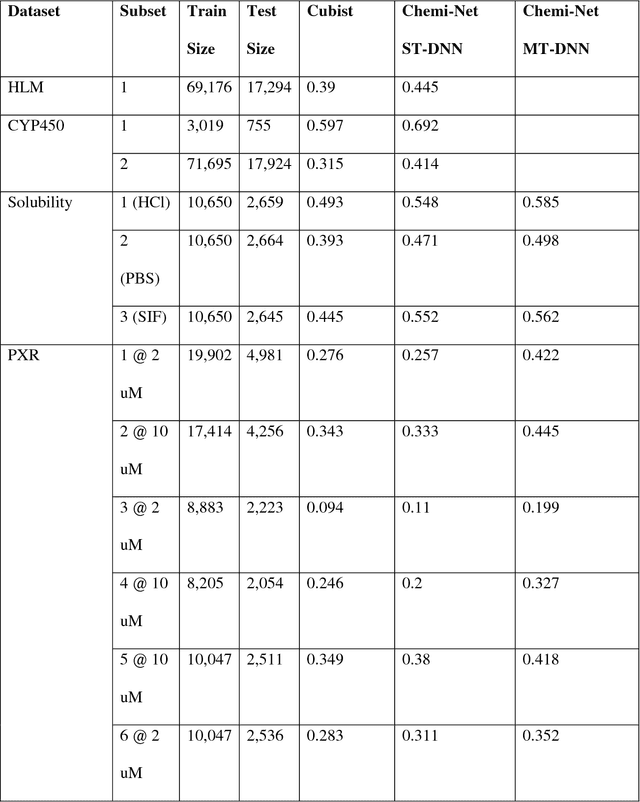
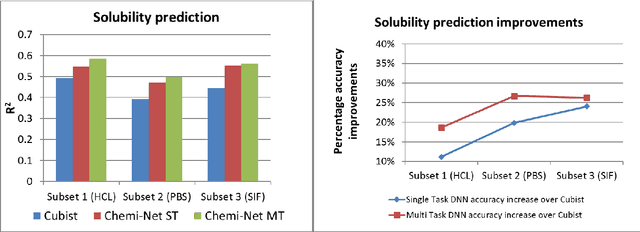

Absorption, distribution, metabolism, and excretion (ADME) studies are critical for drug discovery. Conventionally, these tasks, together with other chemical property predictions, rely on domain-specific feature descriptors, or fingerprints. Following the recent success of neural networks, we developed Chemi-Net, a completely data-driven, domain knowledge-free, deep learning method for ADME property prediction. To compare the relative performance of Chemi-Net with Cubist, one of the popular machine learning programs used by Amgen, a large-scale ADME property prediction study was performed on-site at Amgen. The results showed that our deep neural network method improved current methods by a large margin. We foresee that the significantly increased accuracy of ADME prediction seen with Chemi-Net over Cubist will greatly accelerate drug discovery.