 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTransformers for molecular property prediction: Lessons learned from the past five years
Apr 05, 2024



Molecular Property Prediction (MPP) is vital for drug discovery, crop protection, and environmental science. Over the last decades, diverse computational techniques have been developed, from using simple physical and chemical properties and molecular fingerprints in statistical models and classical machine learning to advanced deep learning approaches. In this review, we aim to distill insights from current research on employing transformer models for MPP. We analyze the currently available models and explore key questions that arise when training and fine-tuning a transformer model for MPP. These questions encompass the choice and scale of the pre-training data, optimal architecture selections, and promising pre-training objectives. Our analysis highlights areas not yet covered in current research, inviting further exploration to enhance the field's understanding. Additionally, we address the challenges in comparing different models, emphasizing the need for standardized data splitting and robust statistical analysis.
Are Learned Molecular Representations Ready For Prime Time?
Apr 02, 2019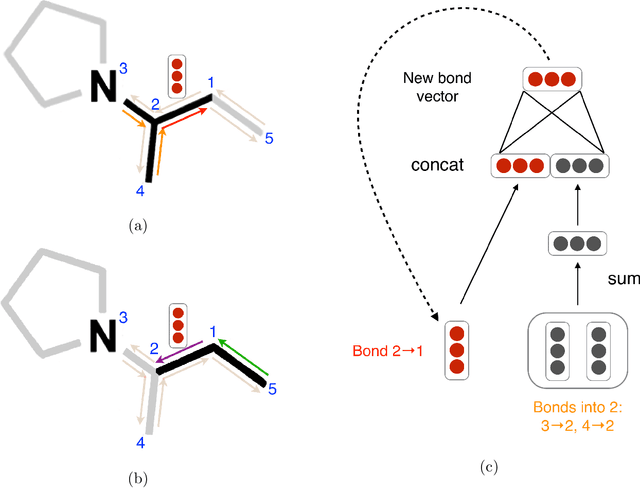


Advancements in neural machinery have led to a wide range of algorithmic solutions for molecular property prediction. Two classes of models in particular have yielded promising results: neural networks applied to computed molecular fingerprints or expert-crafted descriptors, and graph convolutional neural networks that construct a learned molecular representation by operating on the graph structure of the molecule. However, recent literature has yet to clearly determine which of these two methods is superior when generalizing to new chemical space. Furthermore, prior research has rarely examined these new models in industry research settings in comparison to existing employed models. In this paper, we benchmark models extensively on 19 public and 15 proprietary industrial datasets spanning a wide variety of chemical endpoints. In addition, we introduce a graph convolutional model that consistently outperforms models using fixed molecular descriptors as well as previous graph neural architectures on both public and proprietary datasets. Our empirical findings indicate that while approaches based on these representations have yet to reach the level of experimental reproducibility, our proposed model nevertheless offers significant improvements over models currently used in industrial workflows.