 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeChemiRise: a data-driven retrosynthesis engine
Aug 09, 2021
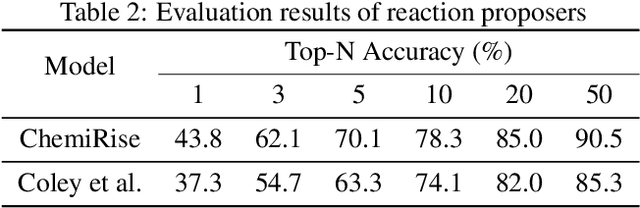

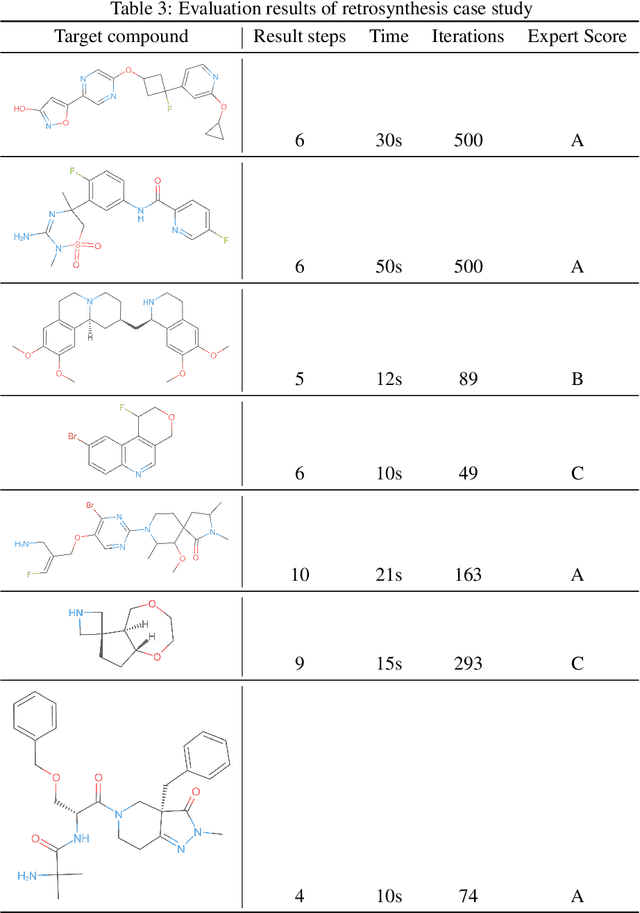
We have developed an end-to-end, retrosynthesis system, named ChemiRise, that can propose complete retrosynthesis routes for organic compounds rapidly and reliably. The system was trained on a processed patent database of over 3 million organic reactions. Experimental reactions were atom-mapped, clustered, and extracted into reaction templates. We then trained a graph convolutional neural network-based one-step reaction proposer using template embeddings and developed a guiding algorithm on the directed acyclic graph (DAG) of chemical compounds to find the best candidate to explore. The atom-mapping algorithm and the one-step reaction proposer were benchmarked against previous studies and showed better results. The final product was demonstrated by retrosynthesis routes reviewed and rated by human experts, showing satisfying functionality and a potential productivity boost in real-life use cases.
Molecular modeling with machine-learned universal potential functions
Mar 06, 2021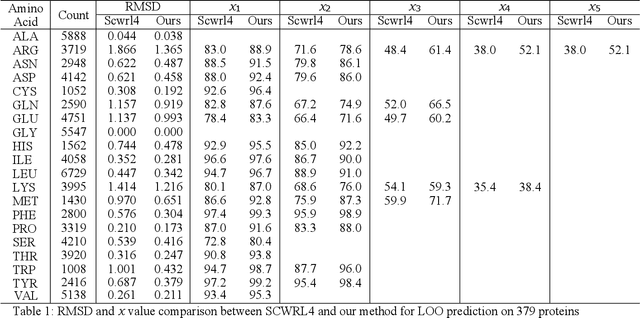

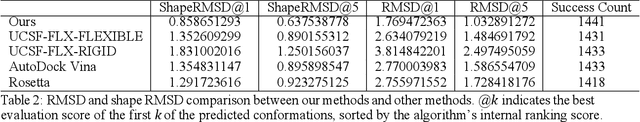
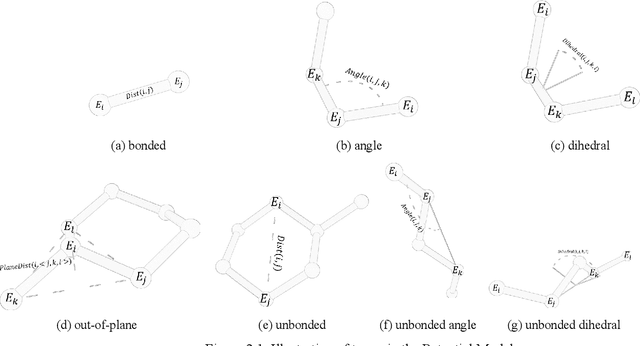
Molecular modeling is an important topic in drug discovery. Decades of research have led to the development of high quality scalable molecular force fields. In this paper, we show that neural networks can be used to train an universal approximator for energy potential functions. By incorporating a fully automated training process we have been able to train smooth, differentiable, and predictive potential functions on large scale crystal structures. A variety of tests have also performed to show the superiority and versatility of the machine-learned model.
Chemi-net: a graph convolutional network for accurate drug property prediction
Mar 21, 2018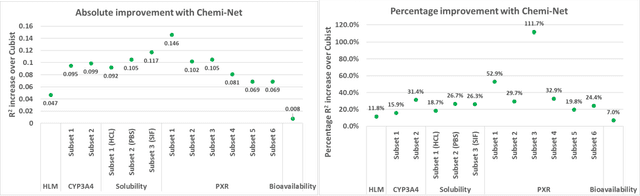
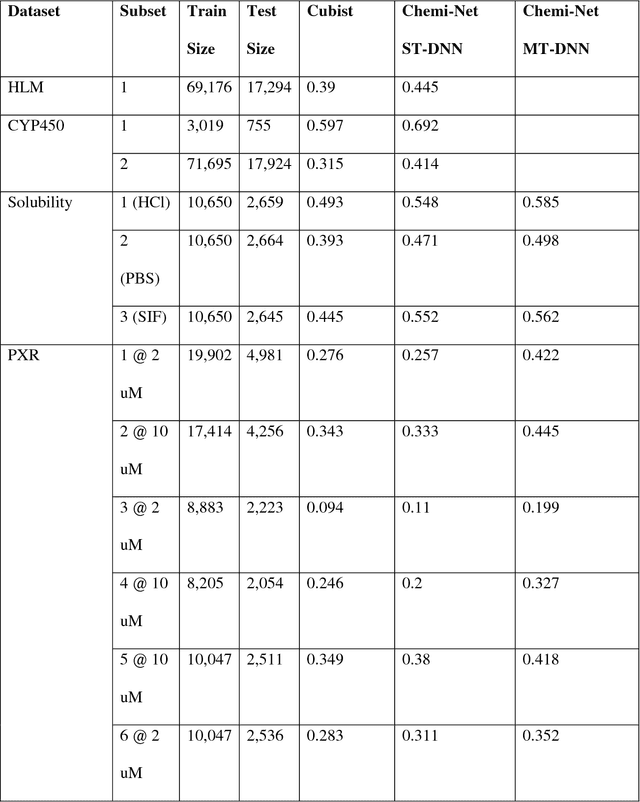
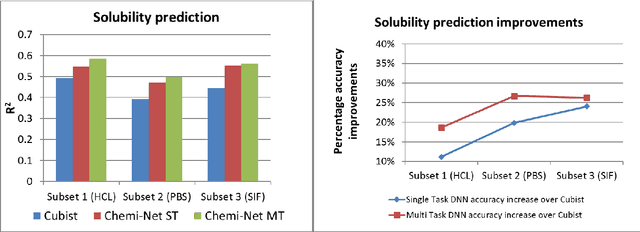

Absorption, distribution, metabolism, and excretion (ADME) studies are critical for drug discovery. Conventionally, these tasks, together with other chemical property predictions, rely on domain-specific feature descriptors, or fingerprints. Following the recent success of neural networks, we developed Chemi-Net, a completely data-driven, domain knowledge-free, deep learning method for ADME property prediction. To compare the relative performance of Chemi-Net with Cubist, one of the popular machine learning programs used by Amgen, a large-scale ADME property prediction study was performed on-site at Amgen. The results showed that our deep neural network method improved current methods by a large margin. We foresee that the significantly increased accuracy of ADME prediction seen with Chemi-Net over Cubist will greatly accelerate drug discovery.
Prediction of amino acid side chain conformation using a deep neural network
Jul 26, 2017
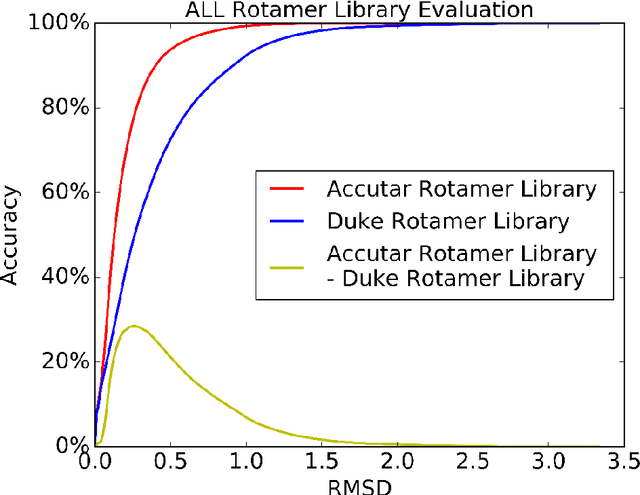
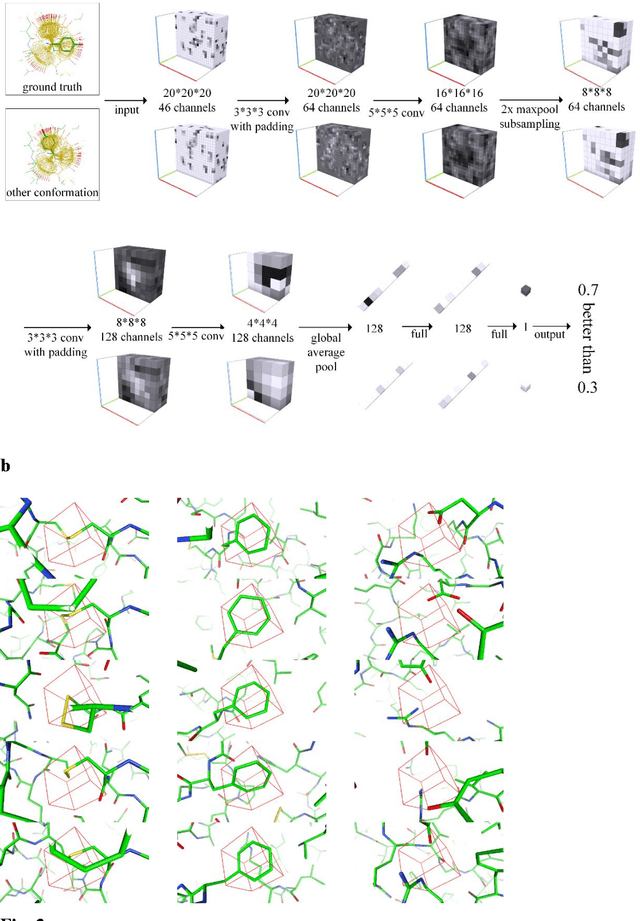
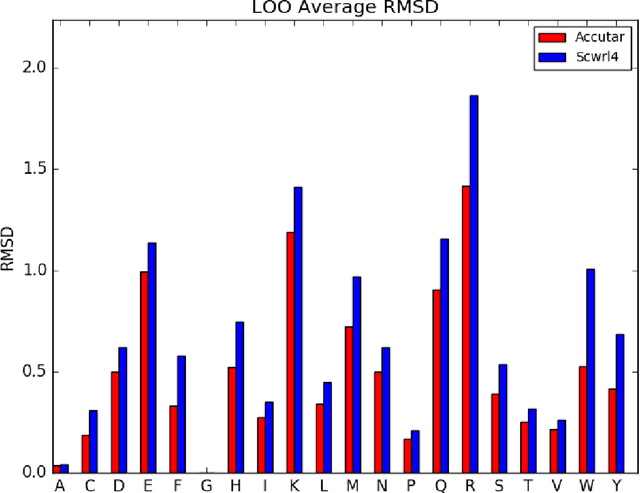
A deep neural network based architecture was constructed to predict amino acid side chain conformation with unprecedented accuracy. Amino acid side chain conformation prediction is essential for protein homology modeling and protein design. Current widely-adopted methods use physics-based energy functions to evaluate side chain conformation. Here, using a deep neural network architecture without physics-based assumptions, we have demonstrated that side chain conformation prediction accuracy can be improved by more than 25%, especially for aromatic residues compared with current standard methods. More strikingly, the prediction method presented here is robust enough to identify individual conformational outliers from high resolution structures in a protein data bank without providing its structural factors. We envisage that our amino acid side chain predictor could be used as a quality check step for future protein structure model validation and many other potential applications such as side chain assignment in Cryo-electron microscopy, crystallography model auto-building, protein folding and small molecule ligand docking.