 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeLUMINA: A Grid Foundation Model for Benchmarking AC Optimal Power Flow Surrogate Learning
May 04, 2026AC optimal power flow (ACOPF) is foundational yet computationally expensive in power grid operations, driving learning-based surrogates for large-scale grid analysis. These surrogates, however, often fail to generalize across network topologies, a critical gap for deployment on grids not seen during training and for routine operational what-if studies. We introduce LUMINA-Bench, a comprehensive benchmark suite for ACOPF surrogate learning covering multi-topology pretraining, transfer, and adaptation. The benchmark evaluates homogeneous and heterogeneous architectures under single- and multi-topology learning settings using unified metrics that capture both predictive accuracy and physics-informed constraint violations. We additionally compare constraint-aware training objectives, including MSE, augmented Lagrangian, and violation-based Lagrangian losses, to characterize accuracy-robustness trade-offs across settings. Data processing, training, and evaluation frameworks are open-sourced as the LUMINA suite to support reproducibility and accelerate future research on feasibility-aware OPF surrogates.
Towards Systematic Generalization for Power Grid Optimization Problems
May 03, 2026AC Optimal Power Flow (ACOPF) and Security-Constrained Unit Commitment (SCUC) are fundamental optimization problems in power system operations. ACOPF serves as the physical backbone of grid simulation and real-time operation, enforcing nonlinear power flow feasibility and network limits, while SCUC represents a core market-level decision process that schedules generation under operational and security constraints. Although these problems share the same underlying transmission network and physical laws, they differ in decision variables and temporal coupling, and prior learning-based approaches address them in isolation, resulting in disjoint models and representations.We propose a learning framework that jointly models ACOPF and SCUC through a shared graph-based backbone that captures grid topology and physical interactions, coupled with task-specific decoders for static and temporal decision-making. Training includes solver supervision with physics-informed objectives to enforce AC feasibility and inter-temporal operational constraints. To evaluate generalization, we assess cross-case transfer on unseen grid topologies for ACOPF and SCUC without retraining, and systematic generalization on the UC-ACOPF problem using unsupervised, physics-based objectives and a power-dispatch consensus mechanism. Experiments across multiple grid scales demonstrate improved performance and transferability relative to existing learning-based baselines, indicating that the model can support learning across heterogeneous power system optimization problems.
LUMINA: Foundation Models for Topology Transferable ACOPF
Mar 04, 2026Foundation models in general promise to accelerate scientific computation by learning reusable representations across problem instances, yet constrained scientific systems, where predictions must satisfy physical laws and safety limits, pose unique challenges that stress conventional training paradigms. We derive design principles for constrained scientific foundation models through systematic investigation of AC optimal power flow (ACOPF), a representative optimization problem in power grid operations where power balance equations and operational constraints are non-negotiable. Through controlled experiments spanning architectures, training objectives, and system diversity, we extract three empirically grounded principles governing scientific foundation model design. These principles characterize three design trade-offs: learning physics-invariant representations while respecting system-specific constraints, optimizing accuracy while ensuring constraint satisfaction, and ensuring reliability in high-impact operating regimes. We present the LUMINA framework, including data processing and training pipelines to support reproducible research on physics-informed, feasibility-aware foundation models across scientific applications.
ICML Topological Deep Learning Challenge 2024: Beyond the Graph Domain
Sep 08, 2024


This paper describes the 2nd edition of the ICML Topological Deep Learning Challenge that was hosted within the ICML 2024 ELLIS Workshop on Geometry-grounded Representation Learning and Generative Modeling (GRaM). The challenge focused on the problem of representing data in different discrete topological domains in order to bridge the gap between Topological Deep Learning (TDL) and other types of structured datasets (e.g. point clouds, graphs). Specifically, participants were asked to design and implement topological liftings, i.e. mappings between different data structures and topological domains --like hypergraphs, or simplicial/cell/combinatorial complexes. The challenge received 52 submissions satisfying all the requirements. This paper introduces the main scope of the challenge, and summarizes the main results and findings.
Large Language Models for Anomaly Detection in Computational Workflows: from Supervised Fine-Tuning to In-Context Learning
Jul 24, 2024



Anomaly detection in computational workflows is critical for ensuring system reliability and security. However, traditional rule-based methods struggle to detect novel anomalies. This paper leverages large language models (LLMs) for workflow anomaly detection by exploiting their ability to learn complex data patterns. Two approaches are investigated: 1) supervised fine-tuning (SFT), where pre-trained LLMs are fine-tuned on labeled data for sentence classification to identify anomalies, and 2) in-context learning (ICL) where prompts containing task descriptions and examples guide LLMs in few-shot anomaly detection without fine-tuning. The paper evaluates the performance, efficiency, generalization of SFT models, and explores zero-shot and few-shot ICL prompts and interpretability enhancement via chain-of-thought prompting. Experiments across multiple workflow datasets demonstrate the promising potential of LLMs for effective anomaly detection in complex executions.
Physics-Informed Heterogeneous Graph Neural Networks for DC Blocker Placement
May 16, 2024



The threat of geomagnetic disturbances (GMDs) to the reliable operation of the bulk energy system has spurred the development of effective strategies for mitigating their impacts. One such approach involves placing transformer neutral blocking devices, which interrupt the path of geomagnetically induced currents (GICs) to limit their impact. The high cost of these devices and the sparsity of transformers that experience high GICs during GMD events, however, calls for a sparse placement strategy that involves high computational cost. To address this challenge, we developed a physics-informed heterogeneous graph neural network (PIHGNN) for solving the graph-based dc-blocker placement problem. Our approach combines a heterogeneous graph neural network (HGNN) with a physics-informed neural network (PINN) to capture the diverse types of nodes and edges in ac/dc networks and incorporates the physical laws of the power grid. We train the PIHGNN model using a surrogate power flow model and validate it using case studies. Results demonstrate that PIHGNN can effectively and efficiently support the deployment of GIC dc-current blockers, ensuring the continued supply of electricity to meet societal demands. Our approach has the potential to contribute to the development of more reliable and resilient power grids capable of withstanding the growing threat that GMDs pose.
Latent Chemical Space Searching for Plug-in Multi-objective Molecule Generation
Apr 10, 2024



Molecular generation, an essential method for identifying new drug structures, has been supported by advancements in machine learning and computational technology. However, challenges remain in multi-objective generation, model adaptability, and practical application in drug discovery. In this study, we developed a versatile 'plug-in' molecular generation model that incorporates multiple objectives related to target affinity, drug-likeness, and synthesizability, facilitating its application in various drug development contexts. We improved the Particle Swarm Optimization (PSO) in the context of drug discoveries, and identified PSO-ENP as the optimal variant for multi-objective molecular generation and optimization through comparative experiments. The model also incorporates a novel target-ligand affinity predictor, enhancing the model's utility by supporting three-dimensional information and improving synthetic feasibility. Case studies focused on generating and optimizing drug-like big marine natural products were performed, underscoring PSO-ENP's effectiveness and demonstrating its considerable potential for practical drug discovery applications.
Self-supervised Learning for Anomaly Detection in Computational Workflows
Oct 02, 2023



Anomaly detection is the task of identifying abnormal behavior of a system. Anomaly detection in computational workflows is of special interest because of its wide implications in various domains such as cybersecurity, finance, and social networks. However, anomaly detection in computational workflows~(often modeled as graphs) is a relatively unexplored problem and poses distinct challenges. For instance, when anomaly detection is performed on graph data, the complex interdependency of nodes and edges, the heterogeneity of node attributes, and edge types must be accounted for. Although the use of graph neural networks can help capture complex inter-dependencies, the scarcity of labeled anomalous examples from workflow executions is still a significant challenge. To address this problem, we introduce an autoencoder-driven self-supervised learning~(SSL) approach that learns a summary statistic from unlabeled workflow data and estimates the normal behavior of the computational workflow in the latent space. In this approach, we combine generative and contrastive learning objectives to detect outliers in the summary statistics. We demonstrate that by estimating the distribution of normal behavior in the latent space, we can outperform state-of-the-art anomaly detection methods on our benchmark datasets.
Orthogonal Gromov-Wasserstein Discrepancy with Efficient Lower Bound
May 12, 2022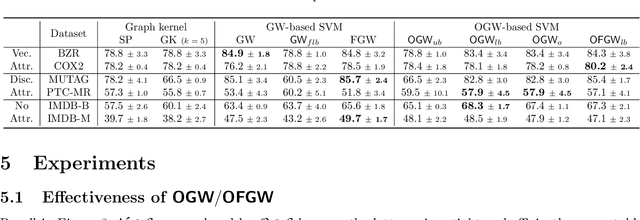
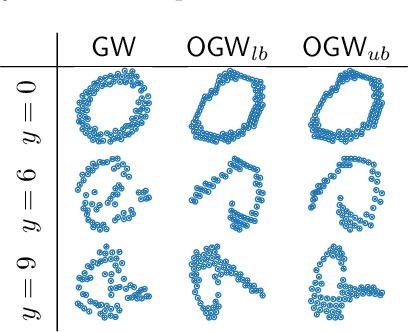
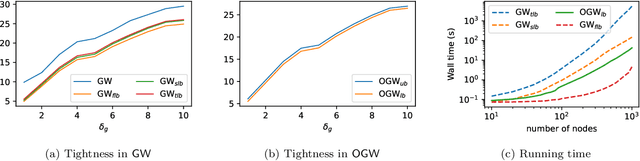
Comparing structured data from possibly different metric-measure spaces is a fundamental task in machine learning, with applications in, e.g., graph classification. The Gromov-Wasserstein (GW) discrepancy formulates a coupling between the structured data based on optimal transportation, tackling the incomparability between different structures by aligning the intra-relational geometries. Although efficient local solvers such as conditional gradient and Sinkhorn are available, the inherent non-convexity still prevents a tractable evaluation, and the existing lower bounds are not tight enough for practical use. To address this issue, we take inspiration from the connection with the quadratic assignment problem, and propose the orthogonal Gromov-Wasserstein (OGW) discrepancy as a surrogate of GW. It admits an efficient and closed-form lower bound with the complexity of $\mathcal{O}(n^3)$, and directly extends to the fused Gromov-Wasserstein (FGW) distance, incorporating node features into the coupling. Extensive experiments on both the synthetic and real-world datasets show the tightness of our lower bounds, and both OGW and its lower bounds efficiently deliver accurate predictions and satisfactory barycenters for graph sets.
Gromov-Wasserstein Discrepancy with Local Differential Privacy for Distributed Structural Graphs
Feb 01, 2022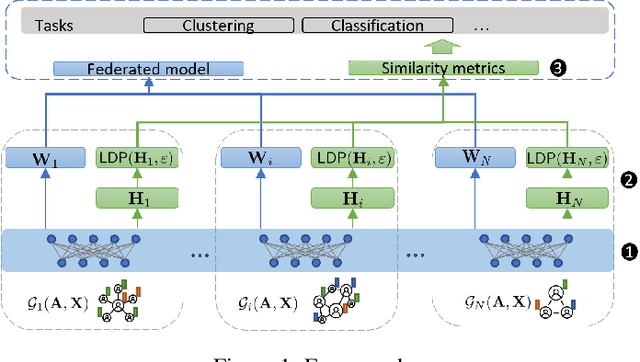
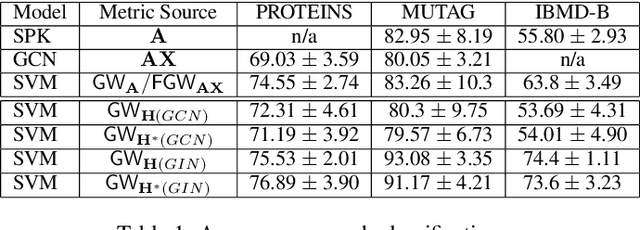

Learning the similarity between structured data, especially the graphs, is one of the essential problems. Besides the approach like graph kernels, Gromov-Wasserstein (GW) distance recently draws big attention due to its flexibility to capture both topological and feature characteristics, as well as handling the permutation invariance. However, structured data are widely distributed for different data mining and machine learning applications. With privacy concerns, accessing the decentralized data is limited to either individual clients or different silos. To tackle these issues, we propose a privacy-preserving framework to analyze the GW discrepancy of node embedding learned locally from graph neural networks in a federated flavor, and then explicitly place local differential privacy (LDP) based on Multi-bit Encoder to protect sensitive information. Our experiments show that, with strong privacy protections guaranteed by the $\varepsilon$-LDP algorithm, the proposed framework not only preserves privacy in graph learning but also presents a noised structural metric under GW distance, resulting in comparable and even better performance in classification and clustering tasks. Moreover, we reason the rationale behind the LDP-based GW distance analytically and empirically.