 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMitosis Detection in the Wild: Multi-Tumor and Context-Aware Generalization in the MIDOG 2025 Challenge
Jun 05, 2026Automated mitosis detection is a well-established task in computational pathology. While previous benchmarks focused on scanner-induced domain shift, clinical "real-world" application requires models to be robust across the vast variance to be expected in the histological landscape. The MItosis DOmain Generalization (MIDOG) 2025 challenge was designed to evaluate algorithmic performance across unprecedented biological and contextual diversity. We curated a test dataset of 365 cases, encompassing 12 distinct human, canine and feline tumor types, digitized across multiple scanning platforms. Moving beyond hand-selected hotspots, the challenge required detection also in random tissue areas (representative of the whole slide detection situation) and challenging areas (areas rich in hard negatives). In the second track, we introduced the classification of atypical mitotic figures (AMFs). There were 18 teams submitting to the detection track, with F1 scores ranging up to 0.740. In the AMF detection track, we had 21 submissions with balanced accuracy values up to 0.908. Our analysis reveals that while most models perform reliably in traditional hotspots, significant performance degradation occurs in challenging ROIs, where false positive rates tripled. Furthermore, performance varied significantly across the 12 tumor types, highlighting "blind spots" in current state-of-the-art architectures when encountering rare or highly pleomorphic malignancies. Moreover, we evaluated the effectiveness of ensembling and found a mean increases of 1.5 and 1.3 percentage points in F1 score and balanced accuracy, respectively. In contrast, TTA showed no relevant improvement. MIDOG 2025 demonstrates that "in the wild" mitosis detection remains a significant hurdle. The transition from hotspot-only evaluation to a multi-contextual framework provides a more realistic proxy for clinical reliability.
Teacher-Student Model for Detecting and Classifying Mitosis in the MIDOG 2025 Challenge
Sep 03, 2025Counting mitotic figures is time-intensive for pathologists and leads to inter-observer variability. Artificial intelligence (AI) promises a solution by automatically detecting mitotic figures while maintaining decision consistency. However, AI tools are susceptible to domain shift, where a significant drop in performance can occur due to differences in the training and testing sets, including morphological diversity between organs, species, and variations in staining protocols. Furthermore, the number of mitoses is much less than the count of normal nuclei, which introduces severely imbalanced data for the detection task. In this work, we formulate mitosis detection as a pixel-level segmentation and propose a teacher-student model that simultaneously addresses mitosis detection (Track 1) and atypical mitosis classification (Track 2). Our method is based on a UNet segmentation backbone that integrates domain generalization modules, namely contrastive representation learning and domain-adversarial training. A teacher-student strategy is employed to generate pixel-level pseudo-masks not only for annotated mitoses and hard negatives but also for normal nuclei, thereby enhancing feature discrimination and improving robustness against domain shift. For the classification task, we introduce a multi-scale CNN classifier that leverages feature maps from the segmentation model within a multi-task learning paradigm. On the preliminary test set, the algorithm achieved an F1 score of 0.7660 in Track 1 and balanced accuracy of 0.8414 in Track 2, demonstrating the effectiveness of integrating segmentation-based detection and classification into a unified framework for robust mitosis analysis.
Domain Adaptation using Silver Standard Masks for Lateral Ventricle Segmentation in FLAIR MRI
Jul 17, 2023



Lateral ventricular volume (LVV) is an important biomarker for clinical investigation. We present the first transfer learning-based LVV segmentation method for fluid-attenuated inversion recovery (FLAIR) MRI. To mitigate covariate shifts between source and target domains, this work proposes an domain adaptation method that optimizes performance on three target datasets. Silver standard (SS) masks were generated from the target domain using a novel conventional image processing ventricular segmentation algorithm and used to supplement the gold standard (GS) data from the source domain, Canadian Atherosclerosis Imaging Network (CAIN). Four models were tested on held-out test sets from four datasets: 1) SS+GS: trained on target SS masks and fine-tuned on source GS masks, 2) GS+SS: trained on source GS masks and fine-tuned on target SS masks, 3) trained on source GS (GS CAIN Only) and 4) trained on target SS masks (SS Only). The SS+GS model had the best and most consistent performance (mean DSC = 0.89, CoV = 0.05) and showed significantly (p < 0.05) higher DSC compared to the GS-only model on three target domains. Results suggest pre-training with noisy labels from the target domain allows the model to adapt to the dataset-specific characteristics and provides robust parameter initialization while fine-tuning with GS masks allows the model to learn detailed features. This method has wide application to other medical imaging problems where labeled data is scarce, and can be used as a per-dataset calibration method to accelerate wide-scale adoption.
Domain Adaptation using Silver Standard Labels for Ki-67 Scoring in Digital Pathology: A Step Closer to Widescale Deployment
Jul 08, 2023



Deep learning systems have been proposed to improve the objectivity and efficiency of Ki- 67 PI scoring. The challenge is that while very accurate, deep learning techniques suffer from reduced performance when applied to out-of-domain data. This is a critical challenge for clinical translation, as models are typically trained using data available to the vendor, which is not from the target domain. To address this challenge, this study proposes a domain adaptation pipeline that employs an unsupervised framework to generate silver standard (pseudo) labels in the target domain, which is used to augment the gold standard (GS) source domain data. Five training regimes were tested on two validated Ki-67 scoring architectures (UV-Net and piNET), (1) SS Only: trained on target silver standard (SS) labels, (2) GS Only: trained on source GS labels, (3) Mixed: trained on target SS and source GS labels, (4) GS+SS: trained on source GS labels and fine-tuned on target SS labels, and our proposed method (5) SS+GS: trained on source SS labels and fine-tuned on source GS labels. The SS+GS method yielded significantly (p < 0.05) higher PI accuracy (95.9%) and more consistent results compared to the GS Only model on target data. Analysis of t-SNE plots showed features learned by the SS+GS models are more aligned for source and target data, resulting in improved generalization. The proposed pipeline provides an efficient method for learning the target distribution without manual annotations, which are time-consuming and costly to generate for medical images. This framework can be applied to any target site as a per-laboratory calibration method, for widescale deployment.
Effect of Intensity Standardization on Deep Learning for WML Segmentation in Multi-Centre FLAIR MRI
Jul 07, 2023Deep learning (DL) methods for white matter lesion (WML) segmentation in MRI suffer a reduction in performance when applied on data from a scanner or centre that is out-of-distribution (OOD) from the training data. This is critical for translation and widescale adoption, since current models cannot be readily applied to data from new institutions. In this work, we evaluate several intensity standardization methods for MRI as a preprocessing step for WML segmentation in multi-centre Fluid-Attenuated Inversion Recovery (FLAIR) MRI. We evaluate a method specifically developed for FLAIR MRI called IAMLAB along with other popular normalization techniques such as White-strip, Nyul and Z-score. We proposed an Ensemble model that combines predictions from each of these models. A skip-connection UNet (SC UNet) was trained on the standardized images, as well as the original data and segmentation performance was evaluated over several dimensions. The training (in-distribution) data consists of a single study, of 60 volumes, and the test (OOD) data is 128 unseen volumes from three clinical cohorts. Results show IAMLAB and Ensemble provide higher WML segmentation performance compared to models from original data or other normalization methods. IAMLAB & Ensemble have the highest dice similarity coefficient (DSC) on the in-distribution data (0.78 & 0.80) and on clinical OOD data. DSC was significantly higher for IAMLAB compared to the original data (p<0.05) for all lesion categories (LL>25mL: 0.77 vs. 0.71; 10mL<= LL<25mL: 0.66 vs. 0.61; LL<10mL: 0.53 vs. 0.52). The IAMLAB and Ensemble normalization methods are mitigating MRI domain shift and are optimal for DL-based WML segmentation in unseen FLAIR data.
MLP-SRGAN: A Single-Dimension Super Resolution GAN using MLP-Mixer
Mar 11, 2023



We propose a novel architecture called MLP-SRGAN, which is a single-dimension Super Resolution Generative Adversarial Network (SRGAN) that utilizes Multi-Layer Perceptron Mixers (MLP-Mixers) along with convolutional layers to upsample in the slice direction. MLP-SRGAN is trained and validated using high resolution (HR) FLAIR MRI from the MSSEG2 challenge dataset. The method was applied to three multicentre FLAIR datasets (CAIN, ADNI, CCNA) of images with low spatial resolution in the slice dimension to examine performance on held-out (unseen) clinical data. Upsampled results are compared to several state-of-the-art SR networks. For images with high resolution (HR) ground truths, peak-signal-to-noise-ratio (PSNR) and structural similarity index (SSIM) are used to measure upsampling performance. Several new structural, no-reference image quality metrics were proposed to quantify sharpness (edge strength), noise (entropy), and blurriness (low frequency information) in the absence of ground truths. Results show MLP-SRGAN results in sharper edges, less blurring, preserves more texture and fine-anatomical detail, with fewer parameters, faster training/evaluation time, and smaller model size than existing methods. Code for MLP-SRGAN training and inference, data generators, models and no-reference image quality metrics will be available at https://github.com/IAMLAB-Ryerson/MLP-SRGAN.
Biomedical image analysis competitions: The state of current participation practice
Dec 16, 2022The number of international benchmarking competitions is steadily increasing in various fields of machine learning (ML) research and practice. So far, however, little is known about the common practice as well as bottlenecks faced by the community in tackling the research questions posed. To shed light on the status quo of algorithm development in the specific field of biomedical imaging analysis, we designed an international survey that was issued to all participants of challenges conducted in conjunction with the IEEE ISBI 2021 and MICCAI 2021 conferences (80 competitions in total). The survey covered participants' expertise and working environments, their chosen strategies, as well as algorithm characteristics. A median of 72% challenge participants took part in the survey. According to our results, knowledge exchange was the primary incentive (70%) for participation, while the reception of prize money played only a minor role (16%). While a median of 80 working hours was spent on method development, a large portion of participants stated that they did not have enough time for method development (32%). 25% perceived the infrastructure to be a bottleneck. Overall, 94% of all solutions were deep learning-based. Of these, 84% were based on standard architectures. 43% of the respondents reported that the data samples (e.g., images) were too large to be processed at once. This was most commonly addressed by patch-based training (69%), downsampling (37%), and solving 3D analysis tasks as a series of 2D tasks. K-fold cross-validation on the training set was performed by only 37% of the participants and only 50% of the participants performed ensembling based on multiple identical models (61%) or heterogeneous models (39%). 48% of the respondents applied postprocessing steps.
Mitosis domain generalization in histopathology images -- The MIDOG challenge
Apr 06, 2022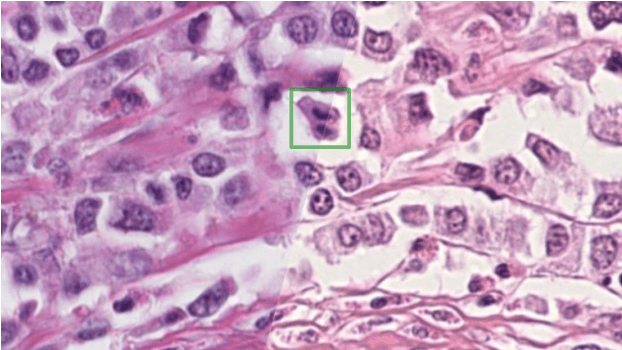
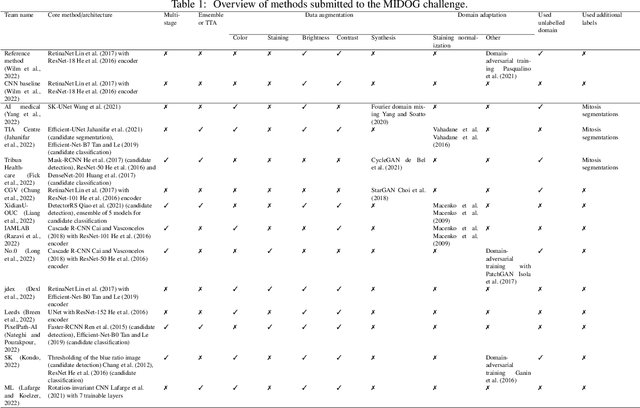
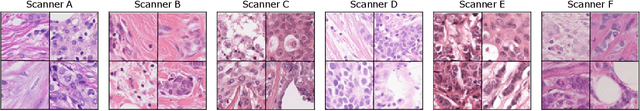
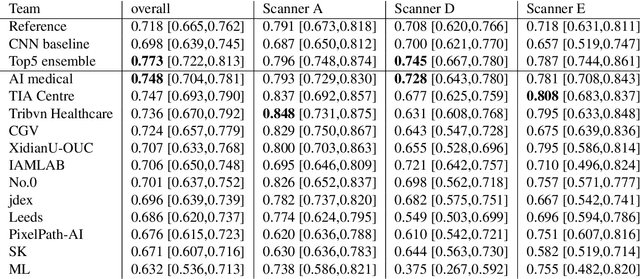
The density of mitotic figures within tumor tissue is known to be highly correlated with tumor proliferation and thus is an important marker in tumor grading. Recognition of mitotic figures by pathologists is known to be subject to a strong inter-rater bias, which limits the prognostic value. State-of-the-art deep learning methods can support the expert in this assessment but are known to strongly deteriorate when applied in a different clinical environment than was used for training. One decisive component in the underlying domain shift has been identified as the variability caused by using different whole slide scanners. The goal of the MICCAI MIDOG 2021 challenge has been to propose and evaluate methods that counter this domain shift and derive scanner-agnostic mitosis detection algorithms. The challenge used a training set of 200 cases, split across four scanning systems. As a test set, an additional 100 cases split across four scanning systems, including two previously unseen scanners, were given. The best approaches performed on an expert level, with the winning algorithm yielding an F_1 score of 0.748 (CI95: 0.704-0.781). In this paper, we evaluate and compare the approaches that were submitted to the challenge and identify methodological factors contributing to better performance.
Preserving Dense Features for Ki67 Nuclei Detection
Nov 10, 2021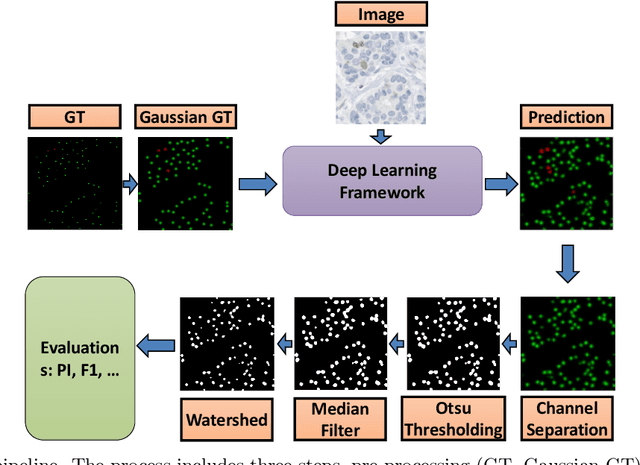
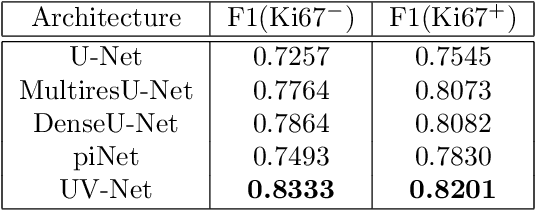
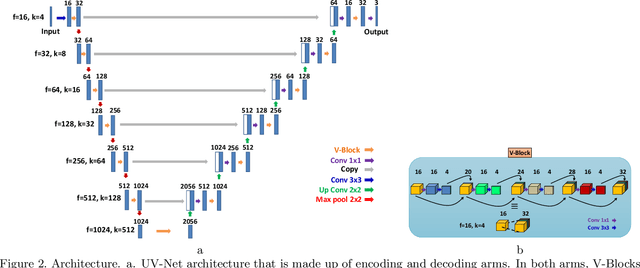
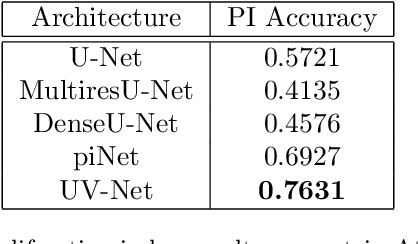
Nuclei detection is a key task in Ki67 proliferation index estimation in breast cancer images. Deep learning algorithms have shown strong potential in nuclei detection tasks. However, they face challenges when applied to pathology images with dense medium and overlapping nuclei since fine details are often diluted or completely lost by early maxpooling layers. This paper introduces an optimized UV-Net architecture, specifically developed to recover nuclear details with high-resolution through feature preservation for Ki67 proliferation index computation. UV-Net achieves an average F1-score of 0.83 on held-out test patch data, while other architectures obtain 0.74-0.79. On tissue microarrays (unseen) test data obtained from multiple centers, UV-Net's accuracy exceeds other architectures by a wide margin, including 9-42\% on Ontario Veterinary College, 7-35\% on Protein Atlas and 0.3-3\% on University Health Network.
Cascade RCNN for MIDOG Challenge
Sep 26, 2021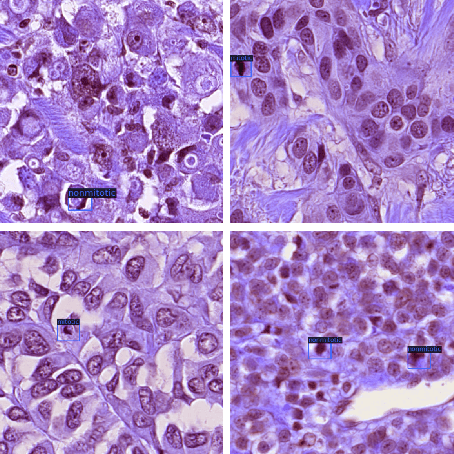
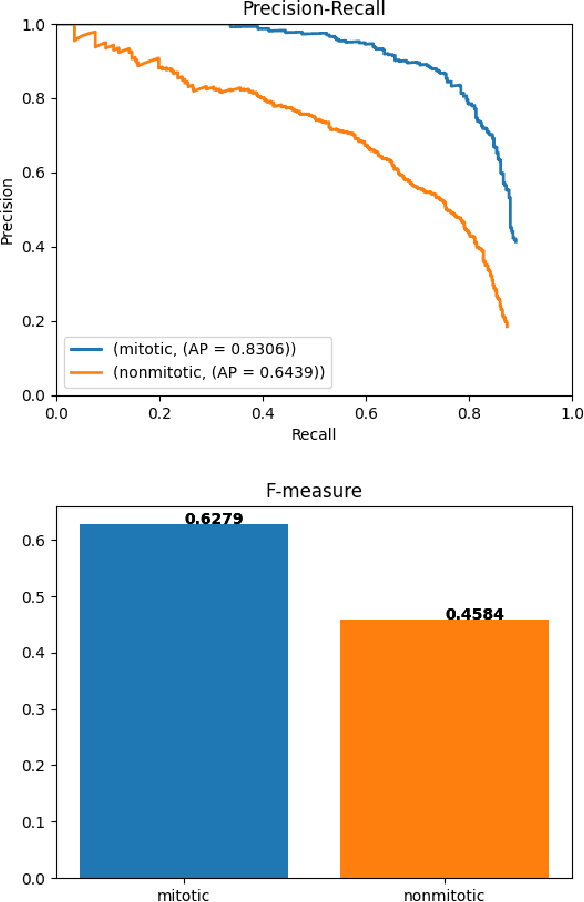
Mitotic counts are one of the key indicators of breast cancer prognosis. However, accurate mitotic cell counting is still a difficult problem and is labourious. Automated methods have been proposed for this task, but are usually dependent on the training images and show poor performance on unseen domains. In this work, we present a multi-stage mitosis detection method based on a Cascade RCNN developed to be sequentially more selective against false positives. On the preliminary test set, the algorithm scores an F1-score of 0.7492.