 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA Deep Learning Framework for Pricing Financial Instruments
Sep 07, 2019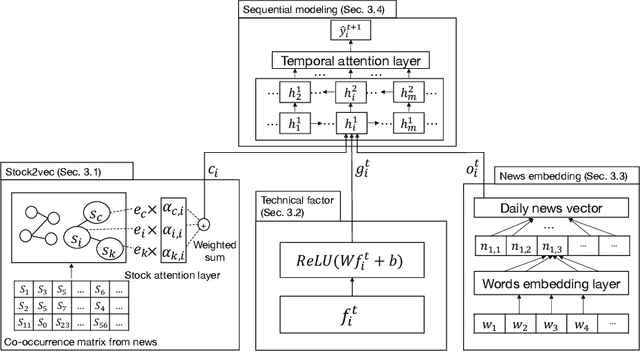
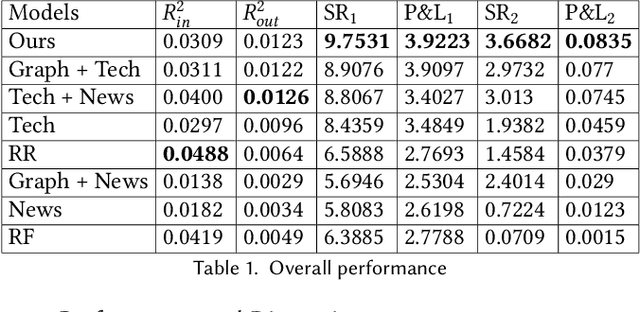

We propose an integrated deep learning architecture for the stock movement prediction. Our architecture simultaneously leverages all available alpha sources. The sources include technical signals, financial news signals, and cross-sectional signals. Our architecture possesses three main properties. First, our architecture eludes overfitting issues. Although we consume a large number of technical signals but has better generalization properties than linear models. Second, our model effectively captures the interactions between signals from different categories. Third, our architecture has low computation cost. We design a graph-based component that extracts cross-sectional interactions which circumvents usage of SVD that's needed in standard models. Experimental results on the real-world stock market show that our approach outperforms the existing baselines. Meanwhile, the results from different trading simulators demonstrate that we can effectively monetize the signals.
DeepScaffold: a comprehensive tool for scaffold-based de novo drug discovery using deep learning
Sep 05, 2019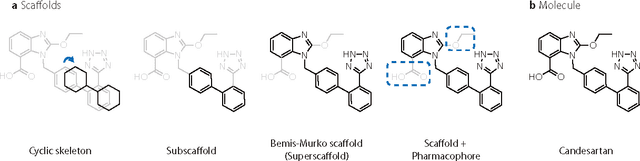
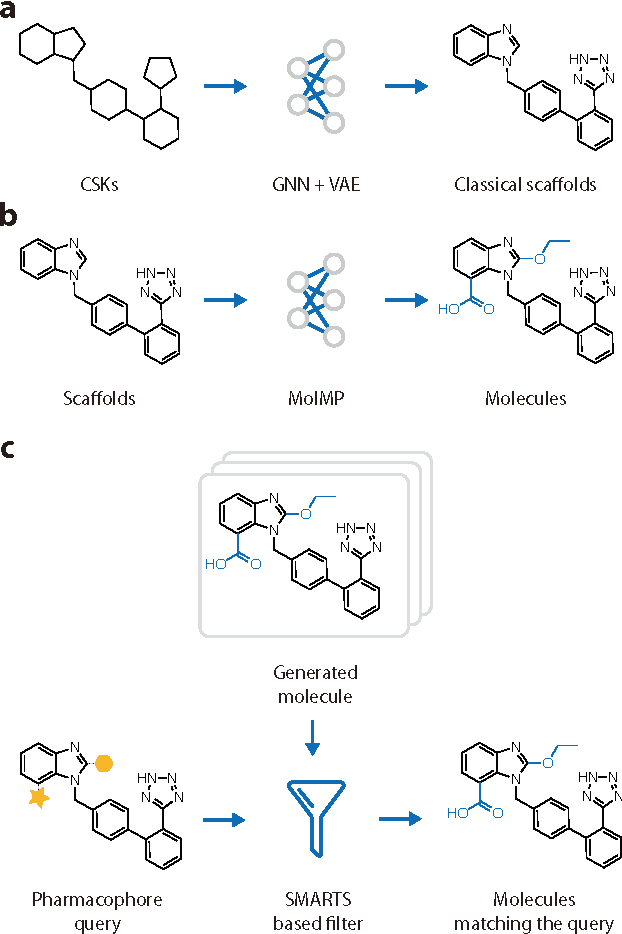
The ultimate goal of drug design is to find novel compounds with desirable pharmacological properties. Designing molecules retaining particular scaffolds as the core structures of the molecules is one of the efficient ways to obtain potential drug candidates with desirable properties. We proposed a scaffold-based molecular generative model for scaffold-based drug discovery, which performs molecule generation based on a wide spectrum of scaffold definitions, including BM-scaffolds, cyclic skeletons, as well as scaffolds with specifications on side-chain properties. The model can generalize the learned chemical rules of adding atoms and bonds to a given scaffold. Furthermore, the generated compounds were evaluated by molecular docking in DRD2 targets and the results demonstrated that this approach can be effectively applied to solve several drug design problems, including the generation of compounds containing a given scaffold and de novo drug design of potential drug candidates with specific docking scores. Finally, a command line interface is created.
Reward Advancement: Transforming Policy under Maximum Causal Entropy Principle
Jul 11, 2019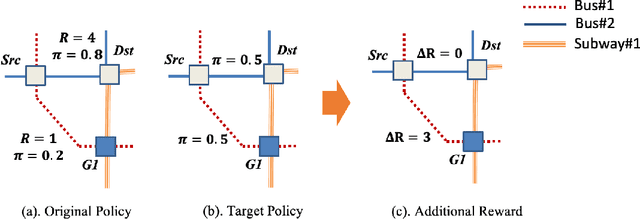
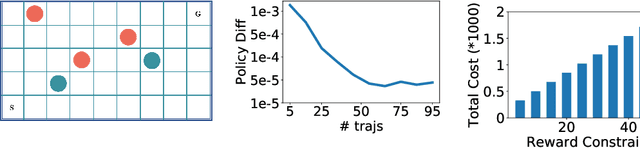
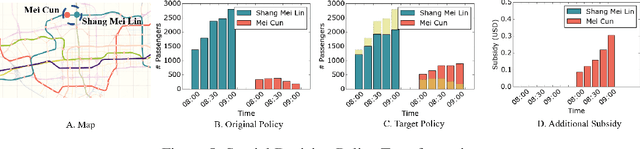
Many real-world human behaviors can be characterized as a sequential decision making processes, such as urban travelers choices of transport modes and routes (Wu et al. 2017). Differing from choices controlled by machines, which in general follows perfect rationality to adopt the policy with the highest reward, studies have revealed that human agents make sub-optimal decisions under bounded rationality (Tao, Rohde, and Corcoran 2014). Such behaviors can be modeled using maximum causal entropy (MCE) principle (Ziebart 2010). In this paper, we define and investigate a general reward trans-formation problem (namely, reward advancement): Recovering the range of additional reward functions that transform the agent's policy from original policy to a predefined target policy under MCE principle. We show that given an MDP and a target policy, there are infinite many additional reward functions that can achieve the desired policy transformation. Moreover, we propose an algorithm to further extract the additional rewards with minimum "cost" to implement the policy transformation.
Adaptive Reduced Rank Regression
May 28, 2019



Low rank regression has proven to be useful in a wide range of forecasting problems. However, in settings with a low signal-to-noise ratio, it is known to suffer from severe overfitting. This paper studies the reduced rank regression problem and presents algorithms with provable generalization guarantees. We use adaptive hard rank-thresholding in two different parts of the data analysis pipeline. First, we consider a low rank projection of the data to eliminate the components that are most likely to be noisy. Second, we perform a standard multivariate linear regression estimator on the data obtained in the first step, and subsequently consider a low-rank projection of the obtained regression matrix. Both thresholding is performed in a data-driven manner and is required to prevent severe overfitting as our lower bounds show. Experimental results show that our approach either outperforms or is competitive with existing baselines.
Towards Non-Parametric Learning to Rank
Jul 09, 2018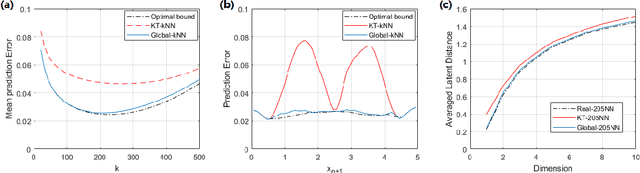
This paper studies a stylized, yet natural, learning-to-rank problem and points out the critical incorrectness of a widely used nearest neighbor algorithm. We consider a model with $n$ agents (users) $\{x_i\}_{i \in [n]}$ and $m$ alternatives (items) $\{y_j\}_{j \in [m]}$, each of which is associated with a latent feature vector. Agents rank items nondeterministically according to the Plackett-Luce model, where the higher the utility of an item to the agent, the more likely this item will be ranked high by the agent. Our goal is to find neighbors of an arbitrary agent or alternative in the latent space. We first show that the Kendall-tau distance based kNN produces incorrect results in our model. Next, we fix the problem by introducing a new algorithm with features constructed from "global information" of the data matrix. Our approach is in sharp contrast to most existing feature engineering methods. Finally, we design another new algorithm identifying similar alternatives. The construction of alternative features can be done using "local information," highlighting the algorithmic difference between finding similar agents and similar alternatives.
Multi-Objective De Novo Drug Design with Conditional Graph Generative Model
Apr 21, 2018

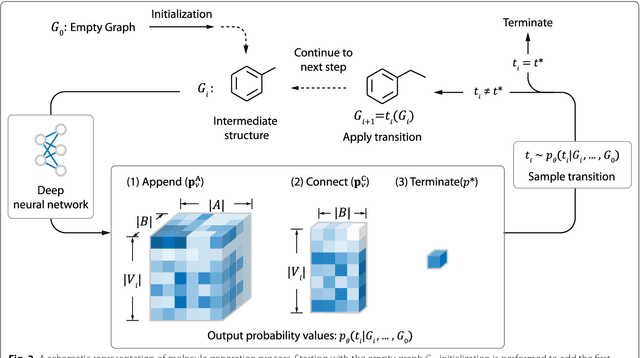
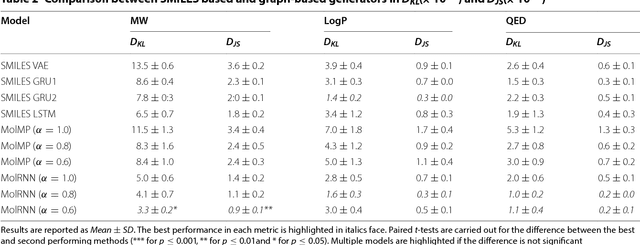
Recently, deep generative models have revealed itself as a promising way of performing de novo molecule design. However, previous research has focused mainly on generating SMILES strings instead of molecular graphs. Although current graph generative models are available, they are often too general and computationally expensive, which restricts their application to molecules with small sizes. In this work, a new de novo molecular design framework is proposed based on a type sequential graph generators that do not use atom level recurrent units. Compared with previous graph generative models, the proposed method is much more tuned for molecule generation and have been scaled up to cover significantly larger molecules in the ChEMBL database. It is shown that the graph-based model outperforms SMILES based models in a variety of metrics, especially in the rate of valid outputs. For the application of drug design tasks, conditional graph generative model is employed. This method offers higher flexibility compared to previous fine-tuning based approach and is suitable for generation based on multiple objectives. This approach is applied to solve several drug design problems, including the generation of compounds containing a given scaffold, generation of compounds with specific drug-likeness and synthetic accessibility requirements, as well as generating dual inhibitors against JNK3 and GSK3$\beta$. Results show high enrichment rates for outputs satisfying the given requirements.
From which world is your graph?
Nov 03, 2017
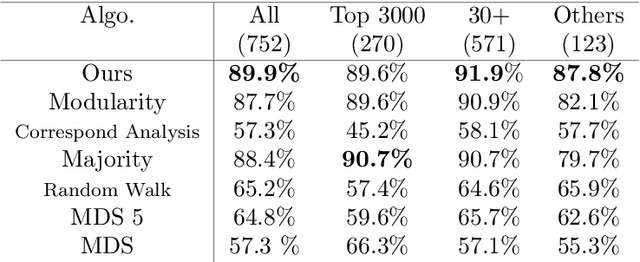

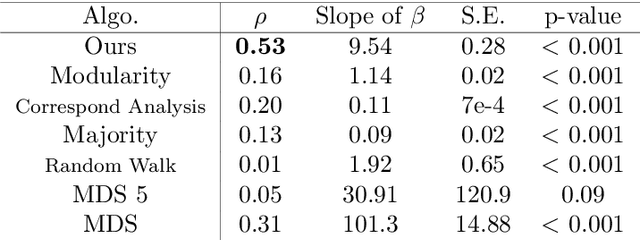
Discovering statistical structure from links is a fundamental problem in the analysis of social networks. Choosing a misspecified model, or equivalently, an incorrect inference algorithm will result in an invalid analysis or even falsely uncover patterns that are in fact artifacts of the model. This work focuses on unifying two of the most widely used link-formation models: the stochastic blockmodel (SBM) and the small world (or latent space) model (SWM). Integrating techniques from kernel learning, spectral graph theory, and nonlinear dimensionality reduction, we develop the first statistically sound polynomial-time algorithm to discover latent patterns in sparse graphs for both models. When the network comes from an SBM, the algorithm outputs a block structure. When it is from an SWM, the algorithm outputs estimates of each node's latent position.
Stock Market Prediction from WSJ: Text Mining via Sparse Matrix Factorization
Jun 27, 2014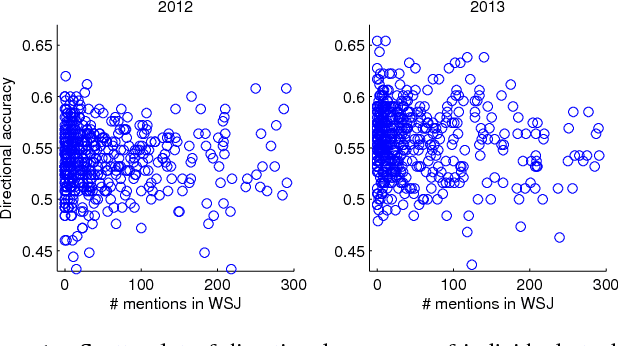
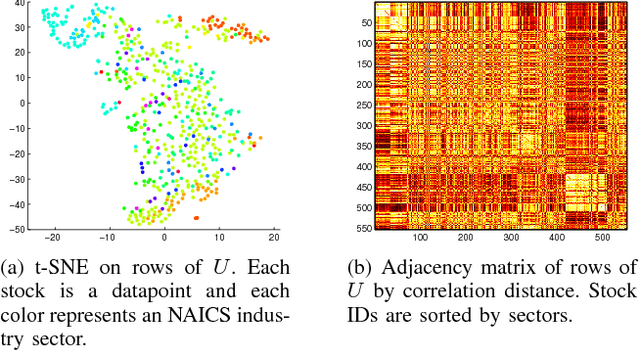
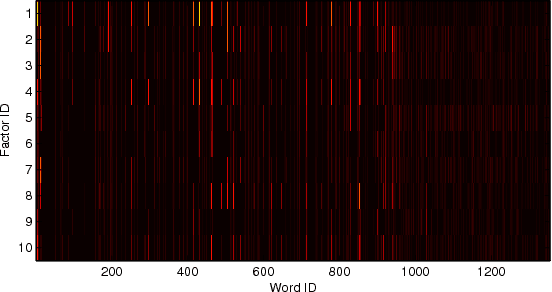
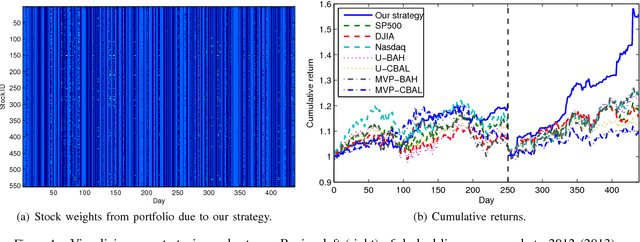
We revisit the problem of predicting directional movements of stock prices based on news articles: here our algorithm uses daily articles from The Wall Street Journal to predict the closing stock prices on the same day. We propose a unified latent space model to characterize the "co-movements" between stock prices and news articles. Unlike many existing approaches, our new model is able to simultaneously leverage the correlations: (a) among stock prices, (b) among news articles, and (c) between stock prices and news articles. Thus, our model is able to make daily predictions on more than 500 stocks (most of which are not even mentioned in any news article) while having low complexity. We carry out extensive backtesting on trading strategies based on our algorithm. The result shows that our model has substantially better accuracy rate (55.7%) compared to many widely used algorithms. The return (56%) and Sharpe ratio due to a trading strategy based on our model are also much higher than baseline indices.
From Black-Scholes to Online Learning: Dynamic Hedging under Adversarial Environments
Jun 23, 2014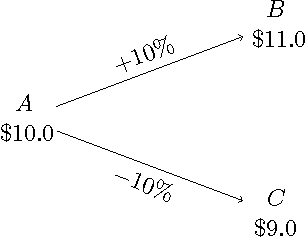
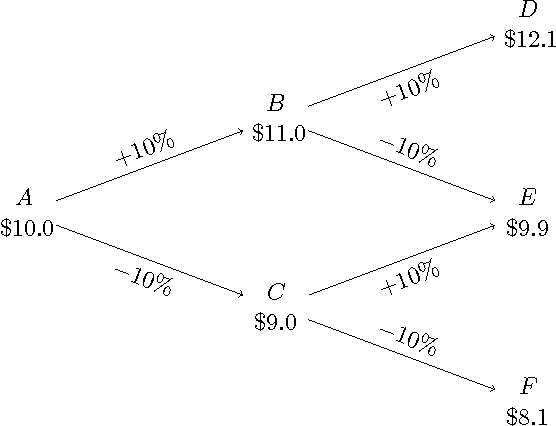
We consider a non-stochastic online learning approach to price financial options by modeling the market dynamic as a repeated game between the nature (adversary) and the investor. We demonstrate that such framework yields analogous structure as the Black-Scholes model, the widely popular option pricing model in stochastic finance, for both European and American options with convex payoffs. In the case of non-convex options, we construct approximate pricing algorithms, and demonstrate that their efficiency can be analyzed through the introduction of an artificial probability measure, in parallel to the so-called risk-neutral measure in the finance literature, even though our framework is completely adversarial. Continuous-time convergence results and extensions to incorporate price jumps are also presented.
Distributed Non-Stochastic Experts
Nov 14, 2012

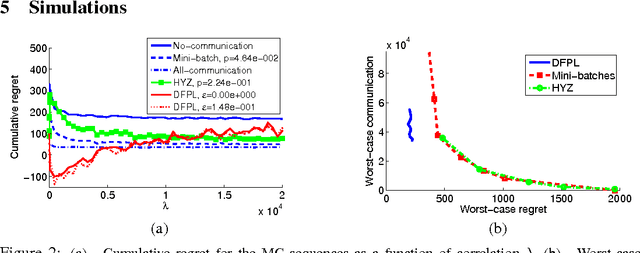
We consider the online distributed non-stochastic experts problem, where the distributed system consists of one coordinator node that is connected to $k$ sites, and the sites are required to communicate with each other via the coordinator. At each time-step $t$, one of the $k$ site nodes has to pick an expert from the set ${1, ..., n}$, and the same site receives information about payoffs of all experts for that round. The goal of the distributed system is to minimize regret at time horizon $T$, while simultaneously keeping communication to a minimum. The two extreme solutions to this problem are: (i) Full communication: This essentially simulates the non-distributed setting to obtain the optimal $O(\sqrt{\log(n)T})$ regret bound at the cost of $T$ communication. (ii) No communication: Each site runs an independent copy : the regret is $O(\sqrt{log(n)kT})$ and the communication is 0. This paper shows the difficulty of simultaneously achieving regret asymptotically better than $\sqrt{kT}$ and communication better than $T$. We give a novel algorithm that for an oblivious adversary achieves a non-trivial trade-off: regret $O(\sqrt{k^{5(1+\epsilon)/6} T})$ and communication $O(T/k^{\epsilon})$, for any value of $\epsilon \in (0, 1/5)$. We also consider a variant of the model, where the coordinator picks the expert. In this model, we show that the label-efficient forecaster of Cesa-Bianchi et al. (2005) already gives us strategy that is near optimal in regret vs communication trade-off.