 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMultimodal Whole Slide Foundation Model for Pathology
Nov 29, 2024



The field of computational pathology has been transformed with recent advances in foundation models that encode histopathology region-of-interests (ROIs) into versatile and transferable feature representations via self-supervised learning (SSL). However, translating these advancements to address complex clinical challenges at the patient and slide level remains constrained by limited clinical data in disease-specific cohorts, especially for rare clinical conditions. We propose TITAN, a multimodal whole slide foundation model pretrained using 335,645 WSIs via visual self-supervised learning and vision-language alignment with corresponding pathology reports and 423,122 synthetic captions generated from a multimodal generative AI copilot for pathology. Without any finetuning or requiring clinical labels, TITAN can extract general-purpose slide representations and generate pathology reports that generalize to resource-limited clinical scenarios such as rare disease retrieval and cancer prognosis. We evaluate TITAN on diverse clinical tasks and find that TITAN outperforms both ROI and slide foundation models across machine learning settings such as linear probing, few-shot and zero-shot classification, rare cancer retrieval and cross-modal retrieval, and pathology report generation.
A General-Purpose Self-Supervised Model for Computational Pathology
Aug 29, 2023



Tissue phenotyping is a fundamental computational pathology (CPath) task in learning objective characterizations of histopathologic biomarkers in anatomic pathology. However, whole-slide imaging (WSI) poses a complex computer vision problem in which the large-scale image resolutions of WSIs and the enormous diversity of morphological phenotypes preclude large-scale data annotation. Current efforts have proposed using pretrained image encoders with either transfer learning from natural image datasets or self-supervised pretraining on publicly-available histopathology datasets, but have not been extensively developed and evaluated across diverse tissue types at scale. We introduce UNI, a general-purpose self-supervised model for pathology, pretrained using over 100 million tissue patches from over 100,000 diagnostic haematoxylin and eosin-stained WSIs across 20 major tissue types, and evaluated on 33 representative CPath clinical tasks in CPath of varying diagnostic difficulties. In addition to outperforming previous state-of-the-art models, we demonstrate new modeling capabilities in CPath such as resolution-agnostic tissue classification, slide classification using few-shot class prototypes, and disease subtyping generalization in classifying up to 108 cancer types in the OncoTree code classification system. UNI advances unsupervised representation learning at scale in CPath in terms of both pretraining data and downstream evaluation, enabling data-efficient AI models that can generalize and transfer to a gamut of diagnostically-challenging tasks and clinical workflows in anatomic pathology.
Pan-Cancer Integrative Histology-Genomic Analysis via Interpretable Multimodal Deep Learning
Aug 04, 2021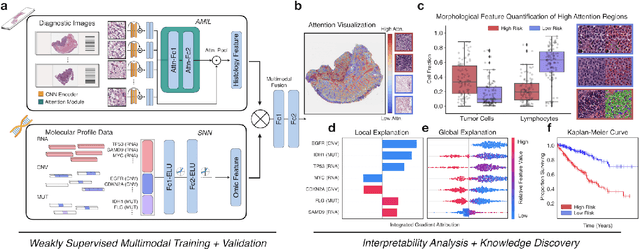
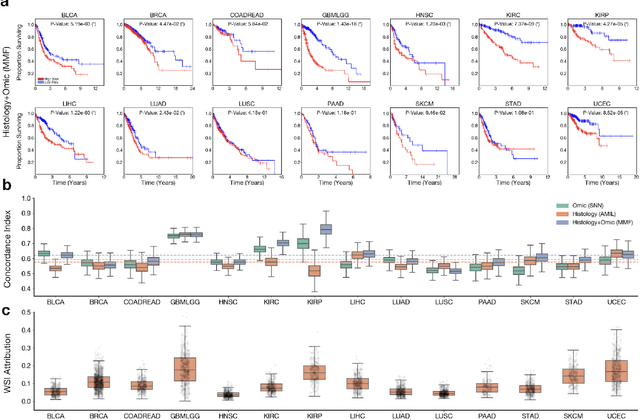
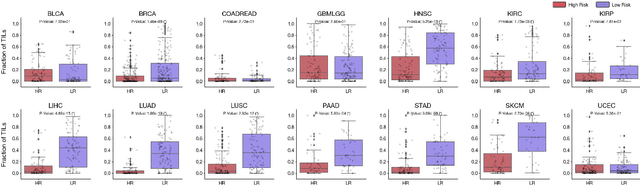
The rapidly emerging field of deep learning-based computational pathology has demonstrated promise in developing objective prognostic models from histology whole slide images. However, most prognostic models are either based on histology or genomics alone and do not address how histology and genomics can be integrated to develop joint image-omic prognostic models. Additionally identifying explainable morphological and molecular descriptors from these models that govern such prognosis is of interest. We used multimodal deep learning to integrate gigapixel whole slide pathology images, RNA-seq abundance, copy number variation, and mutation data from 5,720 patients across 14 major cancer types. Our interpretable, weakly-supervised, multimodal deep learning algorithm is able to fuse these heterogeneous modalities for predicting outcomes and discover prognostic features from these modalities that corroborate with poor and favorable outcomes via multimodal interpretability. We compared our model with unimodal deep learning models trained on histology slides and molecular profiles alone, and demonstrate performance increase in risk stratification on 9 out of 14 cancers. In addition, we analyze morphologic and molecular markers responsible for prognostic predictions across all cancer types. All analyzed data, including morphological and molecular correlates of patient prognosis across the 14 cancer types at a disease and patient level are presented in an interactive open-access database (http://pancancer.mahmoodlab.org) to allow for further exploration and prognostic biomarker discovery. To validate that these model explanations are prognostic, we further analyzed high attention morphological regions in WSIs, which indicates that tumor-infiltrating lymphocyte presence corroborates with favorable cancer prognosis on 9 out of 14 cancer types studied.
Whole Slide Images are 2D Point Clouds: Context-Aware Survival Prediction using Patch-based Graph Convolutional Networks
Jul 27, 2021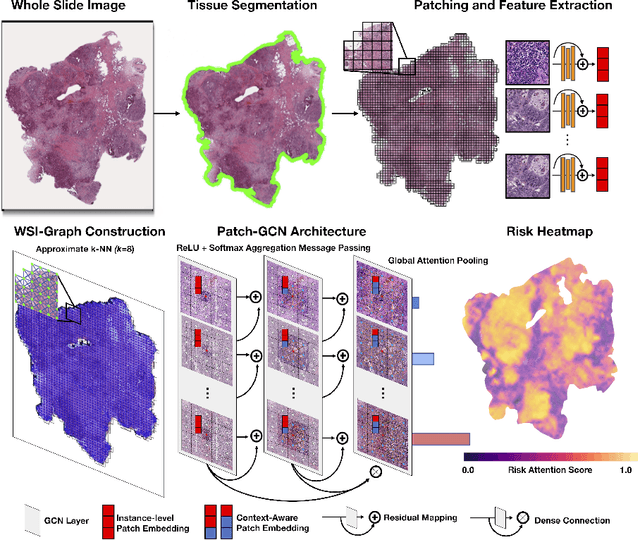
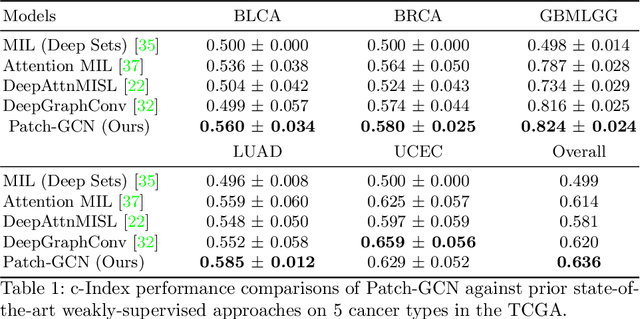
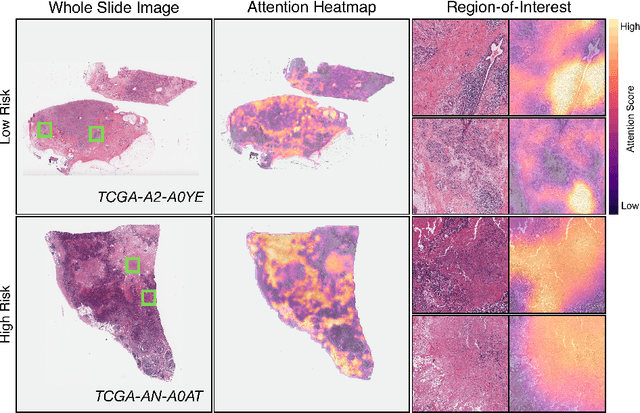
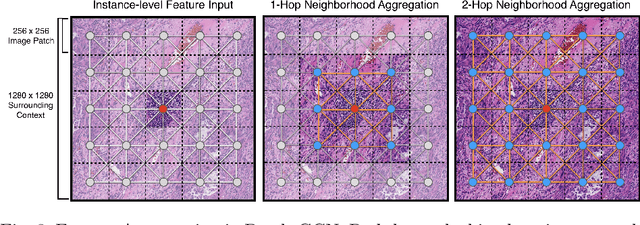
Cancer prognostication is a challenging task in computational pathology that requires context-aware representations of histology features to adequately infer patient survival. Despite the advancements made in weakly-supervised deep learning, many approaches are not context-aware and are unable to model important morphological feature interactions between cell identities and tissue types that are prognostic for patient survival. In this work, we present Patch-GCN, a context-aware, spatially-resolved patch-based graph convolutional network that hierarchically aggregates instance-level histology features to model local- and global-level topological structures in the tumor microenvironment. We validate Patch-GCN with 4,370 gigapixel WSIs across five different cancer types from the Cancer Genome Atlas (TCGA), and demonstrate that Patch-GCN outperforms all prior weakly-supervised approaches by 3.58-9.46%. Our code and corresponding models are publicly available at https://github.com/mahmoodlab/Patch-GCN.
A digital score of tumour-associated stroma infiltrating lymphocytes predicts survival in head and neck squamous cell carcinoma
Apr 16, 2021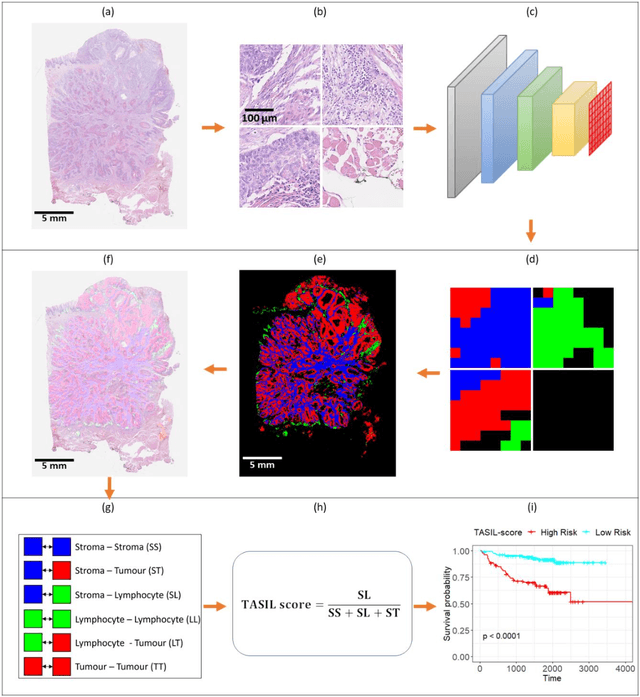
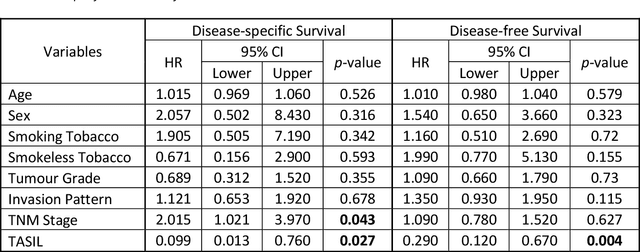
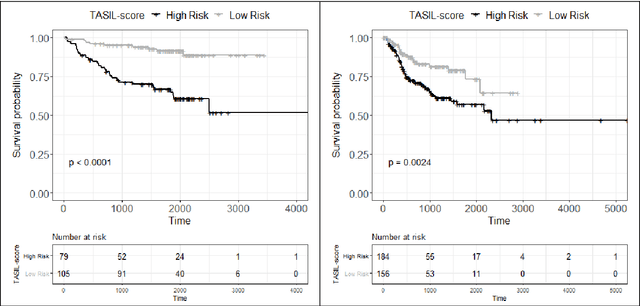
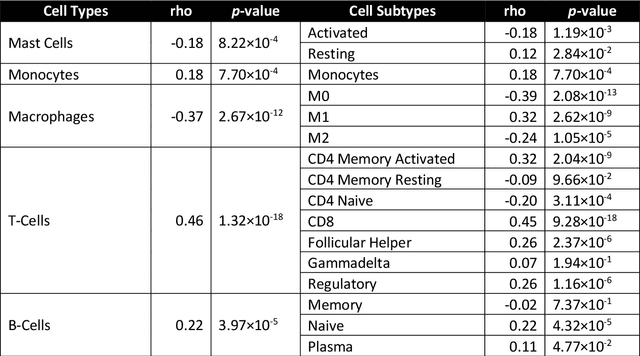
The infiltration of T-lymphocytes in the stroma and tumour is an indication of an effective immune response against the tumour, resulting in better survival. In this study, our aim is to explore the prognostic significance of tumour-associated stroma infiltrating lymphocytes (TASILs) in head and neck squamous cell carcinoma (HNSCC) through an AI based automated method. A deep learning based automated method was employed to segment tumour, stroma and lymphocytes in digitally scanned whole slide images of HNSCC tissue slides. The spatial patterns of lymphocytes and tumour-associated stroma were digitally quantified to compute the TASIL-score. Finally, prognostic significance of the TASIL-score for disease-specific and disease-free survival was investigated with the Cox proportional hazard analysis. Three different cohorts of Haematoxylin & Eosin (H&E) stained tissue slides of HNSCC cases (n=537 in total) were studied, including publicly available TCGA head and neck cancer cases. The TASIL-score carries prognostic significance (p=0.002) for disease-specific survival of HNSCC patients. The TASIL-score also shows a better separation between low- and high-risk patients as compared to the manual TIL scoring by pathologists for both disease-specific and disease-free survival. A positive correlation of TASIL-score with molecular estimates of CD8+ T cells was also found, which is in line with existing findings. To the best of our knowledge, this is the first study to automate the quantification of TASIL from routine H&E slides of head and neck cancer. Our TASIL-score based findings are aligned with the clinical knowledge with the added advantages of objectivity, reproducibility and strong prognostic value. A comprehensive evaluation on large multicentric cohorts is required before the proposed digital score can be adopted in clinical practice.
CGC-Net: Cell Graph Convolutional Network for Grading of Colorectal Cancer Histology Images
Sep 03, 2019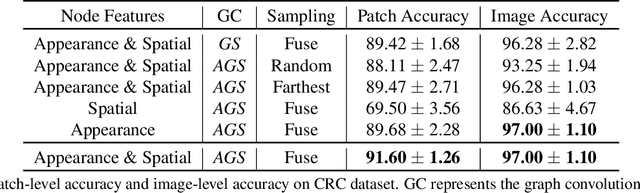
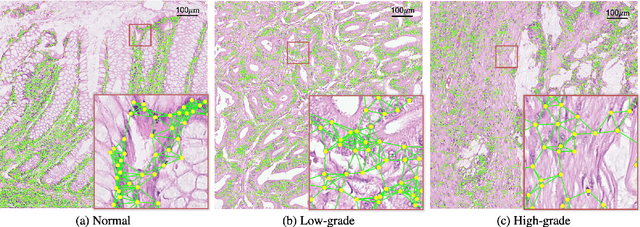
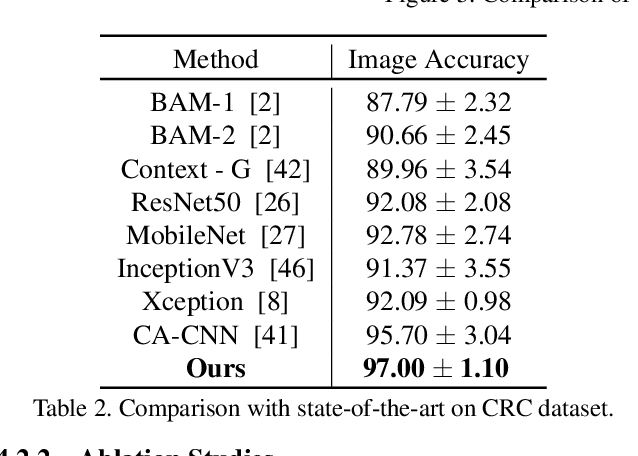
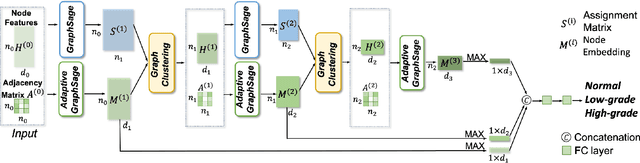
Colorectal cancer (CRC) grading is typically carried out by assessing the degree of gland formation within histology images. To do this, it is important to consider the overall tissue micro-environment by assessing the cell-level information along with the morphology of the gland. However, current automated methods for CRC grading typically utilise small image patches and therefore fail to incorporate the entire tissue micro-architecture for grading purposes. To overcome the challenges of CRC grading, we present a novel cell-graph convolutional neural network (CGC-Net) that converts each large histology image into a graph, where each node is represented by a nucleus within the original image and cellular interactions are denoted as edges between these nodes according to node similarity. The CGC-Net utilises nuclear appearance features in addition to the spatial location of nodes to further boost the performance of the algorithm. To enable nodes to fuse multi-scale information, we introduce Adaptive GraphSage, which is a graph convolution technique that combines multi-level features in a data-driven way. Furthermore, to deal with redundancy in the graph, we propose a sampling technique that removes nodes in areas of dense nuclear activity. We show that modeling the image as a graph enables us to effectively consider a much larger image (around 16$\times$ larger) than traditional patch-based approaches and model the complex structure of the tissue micro-environment. We construct cell graphs with an average of over 3,000 nodes on a large CRC histology image dataset and report state-of-the-art results as compared to recent patch-based as well as contextual patch-based techniques, demonstrating the effectiveness of our method.
Context-Aware Convolutional Neural Network for Grading of Colorectal Cancer Histology Images
Jul 22, 2019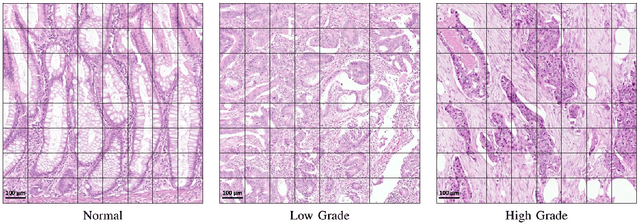
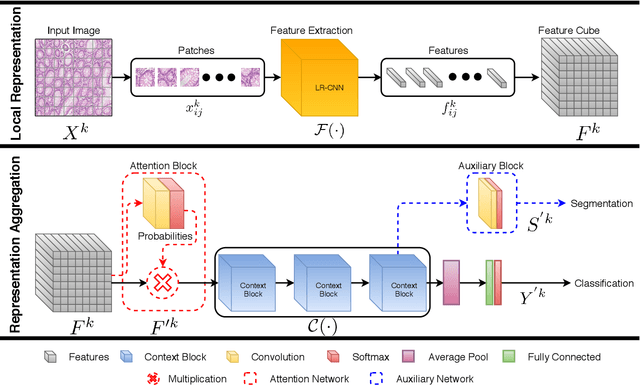
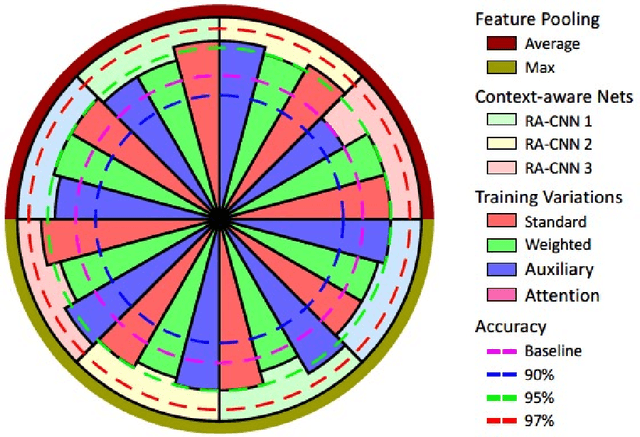

Digital histology images are amenable to the application of convolutional neural network (CNN) for analysis due to the sheer size of pixel data present in them. CNNs are generally used for representation learning from small image patches (e.g. 224x224) extracted from digital histology images due to computational and memory constraints. However, this approach does not incorporate high-resolution contextual information in histology images. We propose a novel way to incorporate larger context by a context-aware neural network based on images with a dimension of 1,792x1,792 pixels. The proposed framework first encodes the local representation of a histology image into high dimensional features then aggregates the features by considering their spatial organization to make a final prediction. The proposed method is evaluated for colorectal cancer grading and breast cancer classification. A comprehensive analysis of some variants of the proposed method is presented. Our method outperformed the traditional patch-based approaches, problem-specific methods, and existing context-based methods quantitatively by a margin of 3.61%. Code and dataset related information is available at this link: https://tia-lab.github.io/Context-Aware-CNN
Methods for Segmentation and Classification of Digital Microscopy Tissue Images
Oct 31, 2018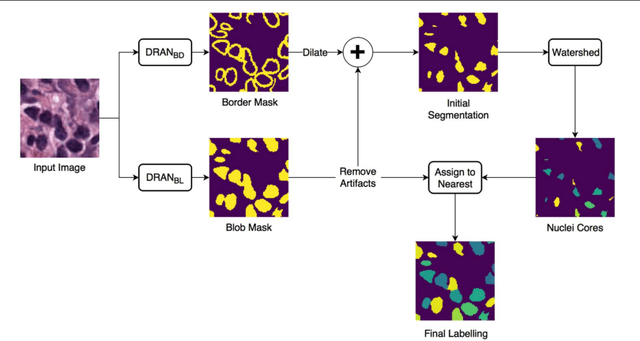

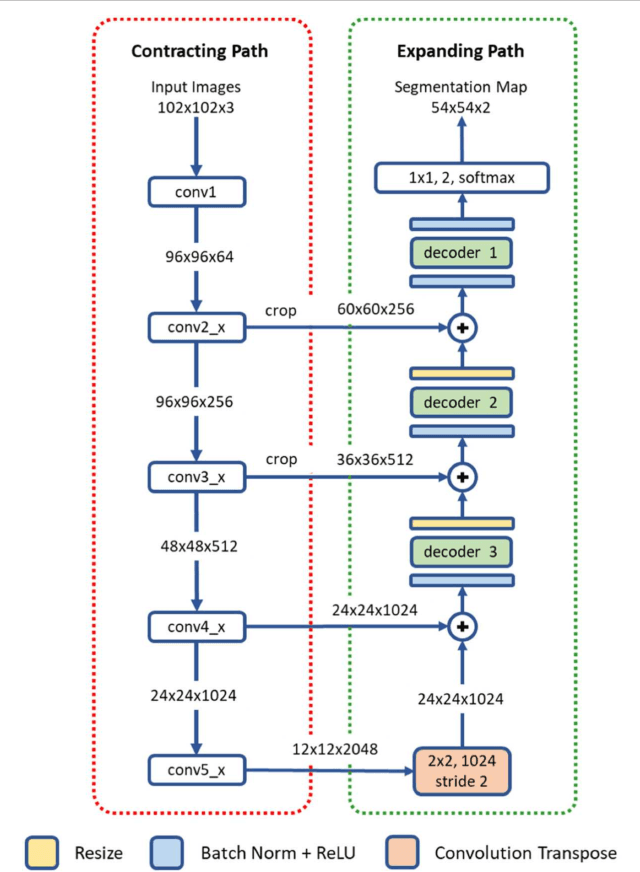
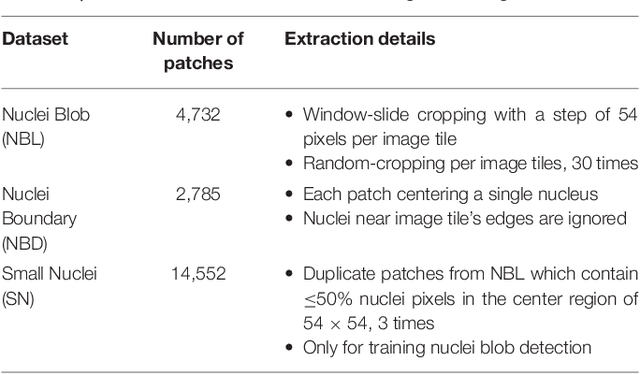
High-resolution microscopy images of tissue specimens provide detailed information about the morphology of normal and diseased tissue. Image analysis of tissue morphology can help cancer researchers develop a better understanding of cancer biology. Segmentation of nuclei and classification of tissue images are two common tasks in tissue image analysis. Development of accurate and efficient algorithms for these tasks is a challenging problem because of the complexity of tissue morphology and tumor heterogeneity. In this paper we present two computer algorithms; one designed for segmentation of nuclei and the other for classification of whole slide tissue images. The segmentation algorithm implements a multiscale deep residual aggregation network to accurately segment nuclear material and then separate clumped nuclei into individual nuclei. The classification algorithm initially carries out patch-level classification via a deep learning method, then patch-level statistical and morphological features are used as input to a random forest regression model for whole slide image classification. The segmentation and classification algorithms were evaluated in the MICCAI 2017 Digital Pathology challenge. The segmentation algorithm achieved an accuracy score of 0.78. The classification algorithm achieved an accuracy score of 0.81.
Micro-Net: A unified model for segmentation of various objects in microscopy images
Apr 22, 2018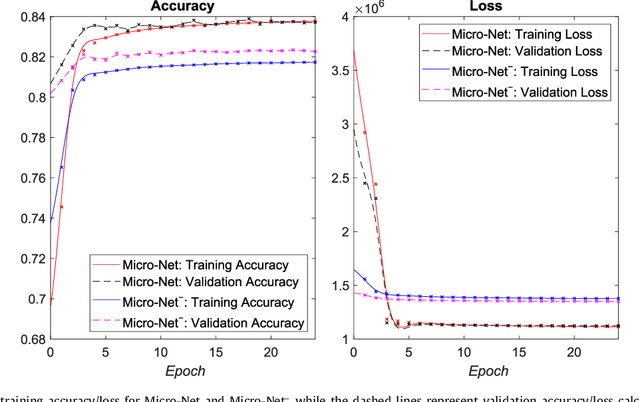
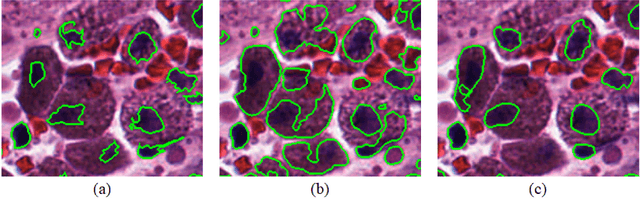
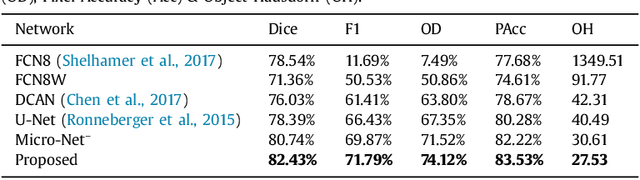
Object segmentation and structure localization are important steps in automated image analysis pipelines for microscopy images. We present a convolution neural network (CNN) based deep learning architecture for segmentation of objects in microscopy images. The proposed network can be used to segment cells, nuclei and glands in fluorescence microscopy and histology images after slight tuning of its parameters. It trains itself at multiple resolutions of the input image, connects the intermediate layers for better localization and context and generates the output using multi-resolution deconvolution filters. The extra convolutional layers which bypass the max-pooling operation allow the network to train for variable input intensities and object size and make it robust to noisy data. We compare our results on publicly available data sets and show that the proposed network outperforms the state-of-the-art.
Context-Aware Learning using Transferable Features for Classification of Breast Cancer Histology Images
Mar 06, 2018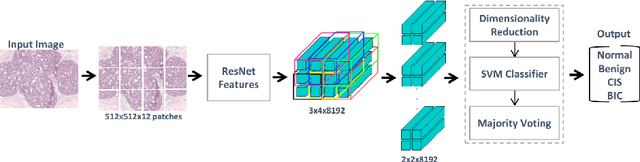
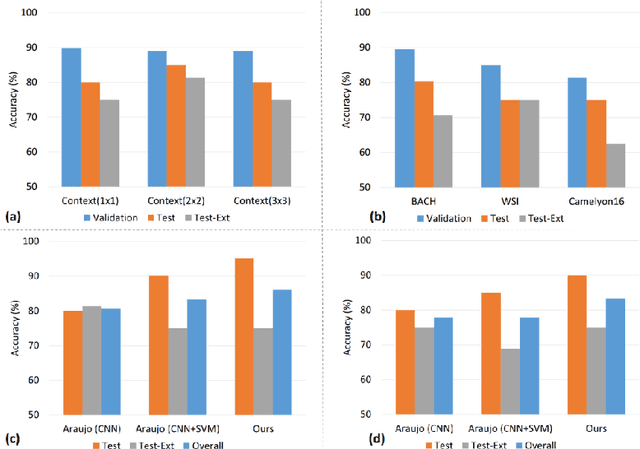
Convolutional neural networks (CNNs) have been recently used for a variety of histology image analysis. However, availability of a large dataset is a major prerequisite for training a CNN which limits its use by the computational pathology community. In previous studies, CNNs have demonstrated their potential in terms of feature generalizability and transferability accompanied with better performance. Considering these traits of CNN, we propose a simple yet effective method which leverages the strengths of CNN combined with the advantages of including contextual information, particularly designed for a small dataset. Our method consists of two main steps: first it uses the activation features of CNN trained for a patch-based classification and then it trains a separate classifier using features of overlapping patches to perform image-based classification using the contextual information. The proposed framework outperformed the state-of-the-art method for breast cancer classification.