 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTowards Agentic Intelligence for Materials Science
Jan 29, 2026The convergence of artificial intelligence and materials science presents a transformative opportunity, but achieving true acceleration in discovery requires moving beyond task-isolated, fine-tuned models toward agentic systems that plan, act, and learn across the full discovery loop. This survey advances a unique pipeline-centric view that spans from corpus curation and pretraining, through domain adaptation and instruction tuning, to goal-conditioned agents interfacing with simulation and experimental platforms. Unlike prior reviews, we treat the entire process as an end-to-end system to be optimized for tangible discovery outcomes rather than proxy benchmarks. This perspective allows us to trace how upstream design choices-such as data curation and training objectives-can be aligned with downstream experimental success through effective credit assignment. To bridge communities and establish a shared frame of reference, we first present an integrated lens that aligns terminology, evaluation, and workflow stages across AI and materials science. We then analyze the field through two focused lenses: From the AI perspective, the survey details LLM strengths in pattern recognition, predictive analytics, and natural language processing for literature mining, materials characterization, and property prediction; from the materials science perspective, it highlights applications in materials design, process optimization, and the acceleration of computational workflows via integration with external tools (e.g., DFT, robotic labs). Finally, we contrast passive, reactive approaches with agentic design, cataloging current contributions while motivating systems that pursue long-horizon goals with autonomy, memory, and tool use. This survey charts a practical roadmap towards autonomous, safety-aware LLM agents aimed at discovering novel and useful materials.
mCLM: A Function-Infused and Synthesis-Friendly Modular Chemical Language Model
May 18, 2025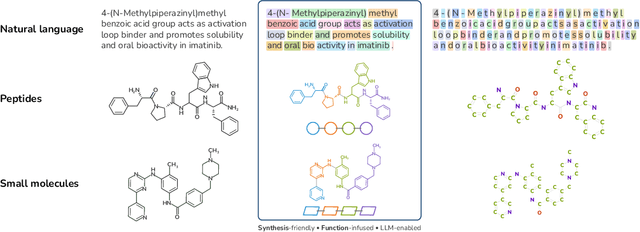
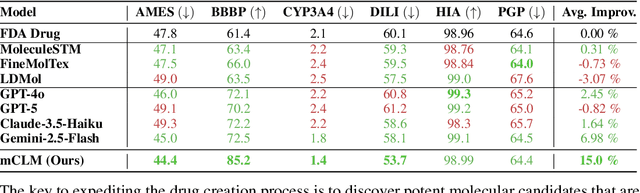
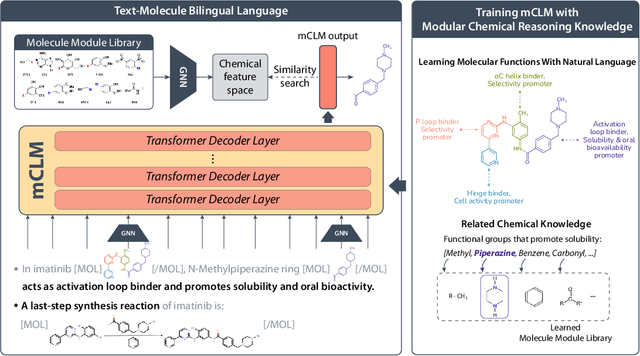
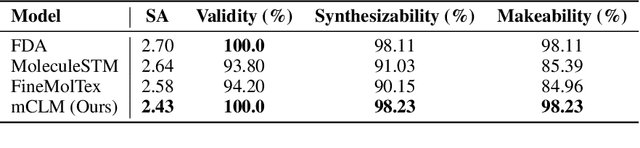
Despite their ability to understand chemical knowledge and accurately generate sequential representations, large language models (LLMs) remain limited in their capacity to propose novel molecules with drug-like properties. In addition, the molecules that LLMs propose can often be challenging to make in the lab. To more effectively enable the discovery of functional small molecules, LLMs need to learn a molecular language. However, LLMs are currently limited by encoding molecules from atoms. In this paper, we argue that just like tokenizing texts into (sub-)word tokens instead of characters, molecules should be decomposed and reassembled at the level of functional building blocks, i.e., parts of molecules that bring unique functions and serve as effective building blocks for real-world automated laboratory synthesis. This motivates us to propose mCLM, a modular Chemical-Language Model tokenizing molecules into building blocks and learning a bilingual language model of both natural language descriptions of functions and molecule building blocks. By reasoning on such functional building blocks, mCLM guarantees to generate efficiently synthesizable molecules thanks to recent progress in block-based chemistry, while also improving the functions of molecules in a principled manner. In experiments on 430 FDA-approved drugs, we find mCLM capable of significantly improving 5 out of 6 chemical functions critical to determining drug potentials. More importantly, mCLM can reason on multiple functions and improve the FDA-rejected drugs (``fallen angels'') over multiple iterations to greatly improve their shortcomings.
FARM: Functional Group-Aware Representations for Small Molecules
Oct 02, 2024



We introduce Functional Group-Aware Representations for Small Molecules (FARM), a novel foundation model designed to bridge the gap between SMILES, natural language, and molecular graphs. The key innovation of FARM lies in its functional group-aware tokenization, which incorporates functional group information directly into the representations. This strategic reduction in tokenization granularity in a way that is intentionally interfaced with key drivers of functional properties (i.e., functional groups) enhances the model's understanding of chemical language, expands the chemical lexicon, more effectively bridging SMILES and natural language, and ultimately advances the model's capacity to predict molecular properties. FARM also represents molecules from two perspectives: by using masked language modeling to capture atom-level features and by employing graph neural networks to encode the whole molecule topology. By leveraging contrastive learning, FARM aligns these two views of representations into a unified molecular embedding. We rigorously evaluate FARM on the MoleculeNet dataset, where it achieves state-of-the-art performance on 10 out of 12 tasks. These results highlight FARM's potential to improve molecular representation learning, with promising applications in drug discovery and pharmaceutical research.
GLaD: Synergizing Molecular Graphs and Language Descriptors for Enhanced Power Conversion Efficiency Prediction in Organic Photovoltaic Devices
May 23, 2024This paper presents a novel approach for predicting Power Conversion Efficiency (PCE) of Organic Photovoltaic (OPV) devices, called GLaD: synergizing molecular Graphs and Language Descriptors for enhanced PCE prediction. Due to the lack of high-quality experimental data, we collect a dataset consisting of 500 pairs of OPV donor and acceptor molecules along with their corresponding PCE values, which we utilize as the training data for our predictive model. In this low-data regime, GLaD leverages properties learned from large language models (LLMs) pretrained on extensive scientific literature to enrich molecular structural representations, allowing for a multimodal representation of molecules. GLaD achieves precise predictions of PCE, thereby facilitating the synthesis of new OPV molecules with improved efficiency. Furthermore, GLaD showcases versatility, as it applies to a range of molecular property prediction tasks (BBBP, BACE, ClinTox, and SIDER), not limited to those concerning OPV materials. Especially, GLaD proves valuable for tasks in low-data regimes within the chemical space, as it enriches molecular representations by incorporating molecular property descriptions learned from large-scale pretraining. This capability is significant in real-world scientific endeavors like drug and material discovery, where access to comprehensive data is crucial for informed decision-making and efficient exploration of the chemical space.