 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeRhythm: Learning Interactive Whole-Body Control for Dual Humanoids
Mar 03, 2026Realizing interactive whole-body control for multi-humanoid systems is critical for unlocking complex collaborative capabilities in shared environments. Although recent advancements have significantly enhanced the agility of individual robots, bridging the gap to physically coupled multi-humanoid interaction remains challenging, primarily due to severe kinematic mismatches and complex contact dynamics. To address this, we introduce Rhythm, the first unified framework enabling real-world deployment of dual-humanoid systems for complex, physically plausible interactions. Our framework integrates three core components: (1) an Interaction-Aware Motion Retargeting (IAMR) module that generates feasible humanoid interaction references from human data; (2) an Interaction-Guided Reinforcement Learning (IGRL) policy that masters coupled dynamics via graph-based rewards; and (3) a real-world deployment system that enables robust transfer of dual-humanoid interaction. Extensive experiments on physical Unitree G1 robots demonstrate that our framework achieves robust interactive whole-body control, successfully transferring diverse behaviors such as hugging and dancing from simulation to reality.
CMoE: Contrastive Mixture of Experts for Motion Control and Terrain Adaptation of Humanoid Robots
Mar 03, 2026For effective deployment in real-world environments, humanoid robots must autonomously navigate a diverse range of complex terrains with abrupt transitions. While the Vanilla mixture of experts (MoE) framework is theoretically capable of modeling diverse terrain features, in practice, the gating network exhibits nearly uniform expert activations across different terrains, weakening the expert specialization and limiting the model's expressive power. To address this limitation, we introduce CMoE, a novel single-stage reinforcement learning framework that integrates contrastive learning to refine expert activation distributions. By imposing contrastive constraints, CMoE maximizes the consistency of expert activations within the same terrain while minimizing their similarity across different terrains, thereby encouraging experts to specialize in distinct terrain types. We validated our approach on the Unitree G1 humanoid robot through a series of challenging experiments. Results demonstrate that CMoE enables the robot to traverse continuous steps up to 20 cm high and gaps up to 80 cm wide, while achieving robust and natural gait across diverse mixed terrains, surpassing the limits of existing methods. To support further research and foster community development, we release our code publicly.
BioReason: Incentivizing Multimodal Biological Reasoning within a DNA-LLM Model
May 29, 2025



Unlocking deep, interpretable biological reasoning from complex genomic data is a major AI challenge hindering scientific discovery. Current DNA foundation models, despite strong sequence representation, struggle with multi-step reasoning and lack inherent transparent, biologically intuitive explanations. We introduce BioReason, a pioneering architecture that, for the first time, deeply integrates a DNA foundation model with a Large Language Model (LLM). This novel connection enables the LLM to directly process and reason with genomic information as a fundamental input, fostering a new form of multimodal biological understanding. BioReason's sophisticated multi-step reasoning is developed through supervised fine-tuning and targeted reinforcement learning, guiding the system to generate logical, biologically coherent deductions. On biological reasoning benchmarks including KEGG-based disease pathway prediction - where accuracy improves from 88% to 97% - and variant effect prediction, BioReason demonstrates an average 15% performance gain over strong single-modality baselines. BioReason reasons over unseen biological entities and articulates decision-making through interpretable, step-by-step biological traces, offering a transformative approach for AI in biology that enables deeper mechanistic insights and accelerates testable hypothesis generation from genomic data. Data, code, and checkpoints are publicly available at https://github.com/bowang-lab/BioReason
Ctrl-DNA: Controllable Cell-Type-Specific Regulatory DNA Design via Constrained RL
May 26, 2025Designing regulatory DNA sequences that achieve precise cell-type-specific gene expression is crucial for advancements in synthetic biology, gene therapy and precision medicine. Although transformer-based language models (LMs) can effectively capture patterns in regulatory DNA, their generative approaches often struggle to produce novel sequences with reliable cell-specific activity. Here, we introduce Ctrl-DNA, a novel constrained reinforcement learning (RL) framework tailored for designing regulatory DNA sequences with controllable cell-type specificity. By formulating regulatory sequence design as a biologically informed constrained optimization problem, we apply RL to autoregressive genomic LMs, enabling the models to iteratively refine sequences that maximize regulatory activity in targeted cell types while constraining off-target effects. Our evaluation on human promoters and enhancers demonstrates that Ctrl-DNA consistently outperforms existing generative and RL-based approaches, generating high-fitness regulatory sequences and achieving state-of-the-art cell-type specificity. Moreover, Ctrl-DNA-generated sequences capture key cell-type-specific transcription factor binding sites (TFBS), short DNA motifs recognized by regulatory proteins that control gene expression, demonstrating the biological plausibility of the generated sequences.
metasnf: Meta Clustering with Similarity Network Fusion in R
Oct 23, 2024



metasnf is an R package that enables users to apply meta clustering, a method for efficiently searching a broad space of cluster solutions by clustering the solutions themselves, to clustering workflows based on similarity network fusion (SNF). SNF is a multi-modal data integration algorithm commonly used for biomedical subtype discovery. The package also contains functions to assist with cluster visualization, characterization, and validation. This package can help researchers identify SNF-derived cluster solutions that are guided by context-specific utility over context-agnostic measures of quality.
Integrate Any Omics: Towards genome-wide data integration for patient stratification
Jan 15, 2024High-throughput omics profiling advancements have greatly enhanced cancer patient stratification. However, incomplete data in multi-omics integration presents a significant challenge, as traditional methods like sample exclusion or imputation often compromise biological diversity and dependencies. Furthermore, the critical task of accurately classifying new patients with partial omics data into existing subtypes is commonly overlooked. To address these issues, we introduce IntegrAO (Integrate Any Omics), an unsupervised framework for integrating incomplete multi-omics data and classifying new samples. IntegrAO first combines partially overlapping patient graphs from diverse omics sources and utilizes graph neural networks to produce unified patient embeddings. Our systematic evaluation across five cancer cohorts involving six omics modalities demonstrates IntegrAO's robustness to missing data and its accuracy in classifying new samples with partial profiles. An acute myeloid leukemia case study further validates its capability to uncover biological and clinical heterogeneity in incomplete datasets. IntegrAO's ability to handle heterogeneous and incomplete data makes it an essential tool for precision oncology, offering a holistic approach to patient characterization.
Unleashing the Strengths of Unlabeled Data in Pan-cancer Abdominal Organ Quantification: the FLARE22 Challenge
Aug 10, 2023



Quantitative organ assessment is an essential step in automated abdominal disease diagnosis and treatment planning. Artificial intelligence (AI) has shown great potential to automatize this process. However, most existing AI algorithms rely on many expert annotations and lack a comprehensive evaluation of accuracy and efficiency in real-world multinational settings. To overcome these limitations, we organized the FLARE 2022 Challenge, the largest abdominal organ analysis challenge to date, to benchmark fast, low-resource, accurate, annotation-efficient, and generalized AI algorithms. We constructed an intercontinental and multinational dataset from more than 50 medical groups, including Computed Tomography (CT) scans with different races, diseases, phases, and manufacturers. We independently validated that a set of AI algorithms achieved a median Dice Similarity Coefficient (DSC) of 90.0\% by using 50 labeled scans and 2000 unlabeled scans, which can significantly reduce annotation requirements. The best-performing algorithms successfully generalized to holdout external validation sets, achieving a median DSC of 89.5\%, 90.9\%, and 88.3\% on North American, European, and Asian cohorts, respectively. They also enabled automatic extraction of key organ biology features, which was labor-intensive with traditional manual measurements. This opens the potential to use unlabeled data to boost performance and alleviate annotation shortages for modern AI models.
Will Multi-modal Data Improves Few-shot Learning?
Jul 25, 2021
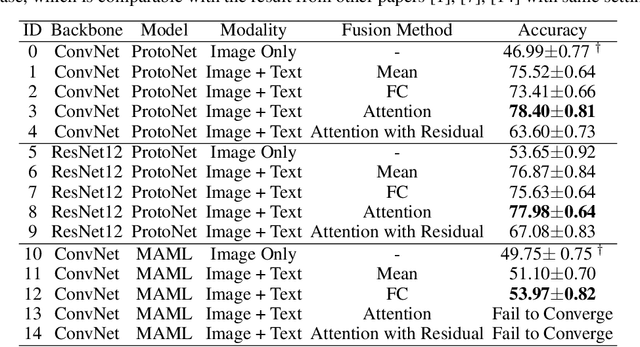
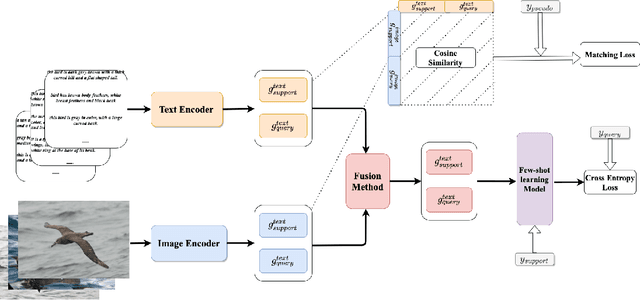

Most few-shot learning models utilize only one modality of data. We would like to investigate qualitatively and quantitatively how much will the model improve if we add an extra modality (i.e. text description of the image), and how it affects the learning procedure. To achieve this goal, we propose four types of fusion method to combine the image feature and text feature. To verify the effectiveness of improvement, we test the fusion methods with two classical few-shot learning models - ProtoNet and MAML, with image feature extractors such as ConvNet and ResNet12. The attention-based fusion method works best, which improves the classification accuracy by a large margin around 30% comparing to the baseline result.