 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDeepAndes: A Self-Supervised Vision Foundation Model for Multi-Spectral Remote Sensing Imagery of the Andes
Apr 28, 2025By mapping sites at large scales using remotely sensed data, archaeologists can generate unique insights into long-term demographic trends, inter-regional social networks, and past adaptations to climate change. Remote sensing surveys complement field-based approaches, and their reach can be especially great when combined with deep learning and computer vision techniques. However, conventional supervised deep learning methods face challenges in annotating fine-grained archaeological features at scale. While recent vision foundation models have shown remarkable success in learning large-scale remote sensing data with minimal annotations, most off-the-shelf solutions are designed for RGB images rather than multi-spectral satellite imagery, such as the 8-band data used in our study. In this paper, we introduce DeepAndes, a transformer-based vision foundation model trained on three million multi-spectral satellite images, specifically tailored for Andean archaeology. DeepAndes incorporates a customized DINOv2 self-supervised learning algorithm optimized for 8-band multi-spectral imagery, marking the first foundation model designed explicitly for the Andes region. We evaluate its image understanding performance through imbalanced image classification, image instance retrieval, and pixel-level semantic segmentation tasks. Our experiments show that DeepAndes achieves superior F1 scores, mean average precision, and Dice scores in few-shot learning scenarios, significantly outperforming models trained from scratch or pre-trained on smaller datasets. This underscores the effectiveness of large-scale self-supervised pre-training in archaeological remote sensing. Codes will be available on https://github.com/geopacha/DeepAndes.
MagNet: Multi-Level Attention Graph Network for Predicting High-Resolution Spatial Transcriptomics
Feb 28, 2025



The rapid development of spatial transcriptomics (ST) offers new opportunities to explore the gene expression patterns within the spatial microenvironment. Current research integrates pathological images to infer gene expression, addressing the high costs and time-consuming processes to generate spatial transcriptomics data. However, as spatial transcriptomics resolution continues to improve, existing methods remain primarily focused on gene expression prediction at low-resolution spot levels. These methods face significant challenges, especially the information bottleneck, when they are applied to high-resolution HD data. To bridge this gap, this paper introduces MagNet, a multi-level attention graph network designed for accurate prediction of high-resolution HD data. MagNet employs cross-attention layers to integrate features from multi-resolution image patches hierarchically and utilizes a GAT-Transformer module to aggregate neighborhood information. By integrating multilevel features, MagNet overcomes the limitations posed by low-resolution inputs in predicting high-resolution gene expression. We systematically evaluated MagNet and existing ST prediction models on both a private spatial transcriptomics dataset and a public dataset at three different resolution levels. The results demonstrate that MagNet achieves state-of-the-art performance at both spot level and high-resolution bin levels, providing a novel methodology and benchmark for future research and applications in high-resolution HD-level spatial transcriptomics. Code is available at https://github.com/Junchao-Zhu/MagNet.
CASC-AI: Consensus-aware Self-corrective AI Agents for Noise Cell Segmentation
Feb 11, 2025Multi-class cell segmentation in high-resolution gigapixel whole slide images (WSI) is crucial for various clinical applications. However, training such models typically requires labor-intensive, pixel-wise annotations by domain experts. Recent efforts have democratized this process by involving lay annotators without medical expertise. However, conventional non-agent-based approaches struggle to handle annotation noise adaptively, as they lack mechanisms to mitigate false positives (FP) and false negatives (FN) at both the image-feature and pixel levels. In this paper, we propose a consensus-aware self-corrective AI agent that leverages the Consensus Matrix to guide its learning process. The Consensus Matrix defines regions where both the AI and annotators agree on cell and non-cell annotations, which are prioritized with stronger supervision. Conversely, areas of disagreement are adaptively weighted based on their feature similarity to high-confidence agreement regions, with more similar regions receiving greater attention. Additionally, contrastive learning is employed to separate features of noisy regions from those of reliable agreement regions by maximizing their dissimilarity. This paradigm enables the AI to iteratively refine noisy labels, enhancing its robustness. Validated on one real-world lay-annotated cell dataset and two simulated noisy datasets, our method demonstrates improved segmentation performance, effectively correcting FP and FN errors and showcasing its potential for training robust models on noisy datasets. The official implementation and cell annotations are publicly available at https://github.com/ddrrnn123/CASC-AI.
KPIs 2024 Challenge: Advancing Glomerular Segmentation from Patch- to Slide-Level
Feb 11, 2025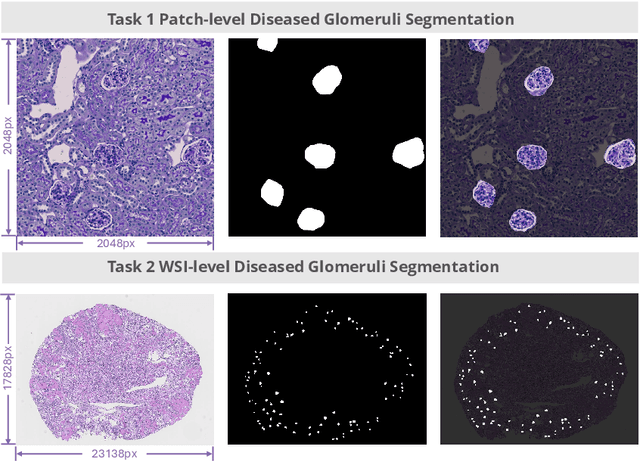
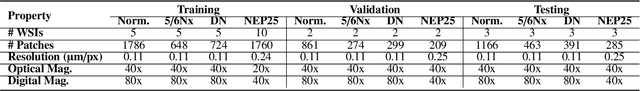
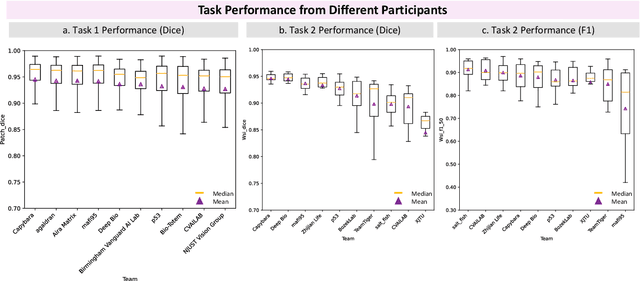

Chronic kidney disease (CKD) is a major global health issue, affecting over 10% of the population and causing significant mortality. While kidney biopsy remains the gold standard for CKD diagnosis and treatment, the lack of comprehensive benchmarks for kidney pathology segmentation hinders progress in the field. To address this, we organized the Kidney Pathology Image Segmentation (KPIs) Challenge, introducing a dataset that incorporates preclinical rodent models of CKD with over 10,000 annotated glomeruli from 60+ Periodic Acid Schiff (PAS)-stained whole slide images. The challenge includes two tasks, patch-level segmentation and whole slide image segmentation and detection, evaluated using the Dice Similarity Coefficient (DSC) and F1-score. By encouraging innovative segmentation methods that adapt to diverse CKD models and tissue conditions, the KPIs Challenge aims to advance kidney pathology analysis, establish new benchmarks, and enable precise, large-scale quantification for disease research and diagnosis.
Towards Fine-grained Renal Vasculature Segmentation: Full-Scale Hierarchical Learning with FH-Seg
Feb 07, 2025Accurate fine-grained segmentation of the renal vasculature is critical for nephrological analysis, yet it faces challenges due to diverse and insufficiently annotated images. Existing methods struggle to accurately segment intricate regions of the renal vasculature, such as the inner and outer walls, arteries and lesions. In this paper, we introduce FH-Seg, a Full-scale Hierarchical Learning Framework designed for comprehensive segmentation of the renal vasculature. Specifically, FH-Seg employs full-scale skip connections that merge detailed anatomical information with contextual semantics across scales, effectively bridging the gap between structural and pathological contexts. Additionally, we implement a learnable hierarchical soft attention gates to adaptively reduce interference from non-core information, enhancing the focus on critical vascular features. To advance research on renal pathology segmentation, we also developed a Large Renal Vasculature (LRV) dataset, which contains 16,212 fine-grained annotated images of 5,600 renal arteries. Extensive experiments on the LRV dataset demonstrate FH-Seg's superior accuracies (71.23% Dice, 73.06% F1), outperforming Omni-Seg by 2.67 and 2.13 percentage points respectively. Code is available at: https://github.com/hrlblab/FH-seg.
Enhanced Feature-based Image Stitching for Endoscopic Videos in Pediatric Eosinophilic Esophagitis
Feb 06, 2025



Video endoscopy represents a major advance in the investigation of gastrointestinal diseases. Reviewing endoscopy videos often involves frequent adjustments and reorientations to piece together a complete view, which can be both time-consuming and prone to errors. Image stitching techniques address this issue by providing a continuous and complete visualization of the examined area. However, endoscopic images, particularly those of the esophagus, present unique challenges. The smooth surface, lack of distinct feature points, and non-horizontal orientation complicate the stitching process, rendering traditional feature-based methods often ineffective for these types of images. In this paper, we propose a novel preprocessing pipeline designed to enhance endoscopic image stitching through advanced computational techniques. Our approach converts endoscopic video data into continuous 2D images by following four key steps: (1) keyframe selection, (2) image rotation adjustment to correct distortions, (3) surface unwrapping using polar coordinate transformation to generate a flat image, and (4) feature point matching enhanced by Adaptive Histogram Equalization for improved feature detection. We evaluate stitching quality through the assessment of valid feature point match pairs. Experiments conducted on 20 pediatric endoscopy videos demonstrate that our method significantly improves image alignment and stitching quality compared to traditional techniques, laying a robust foundation for more effective panoramic image creation.
Expanding Training Data for Endoscopic Phenotyping of Eosinophilic Esophagitis
Feb 06, 2025Eosinophilic esophagitis (EoE) is a chronic esophageal disorder marked by eosinophil-dominated inflammation. Diagnosing EoE usually involves endoscopic inspection of the esophageal mucosa and obtaining esophageal biopsies for histologic confirmation. Recent advances have seen AI-assisted endoscopic imaging, guided by the EREFS system, emerge as a potential alternative to reduce reliance on invasive histological assessments. Despite these advancements, significant challenges persist due to the limited availability of data for training AI models - a common issue even in the development of AI for more prevalent diseases. This study seeks to improve the performance of deep learning-based EoE phenotype classification by augmenting our training data with a diverse set of images from online platforms, public datasets, and electronic textbooks increasing our dataset from 435 to 7050 images. We utilized the Data-efficient Image Transformer for image classification and incorporated attention map visualizations to boost interpretability. The findings show that our expanded dataset and model enhancements improved diagnostic accuracy, robustness, and comprehensive analysis, enhancing patient outcomes.
PySpatial: A High-Speed Whole Slide Image Pathomics Toolkit
Jan 10, 2025



Whole Slide Image (WSI) analysis plays a crucial role in modern digital pathology, enabling large-scale feature extraction from tissue samples. However, traditional feature extraction pipelines based on tools like CellProfiler often involve lengthy workflows, requiring WSI segmentation into patches, feature extraction at the patch level, and subsequent mapping back to the original WSI. To address these challenges, we present PySpatial, a high-speed pathomics toolkit specifically designed for WSI-level analysis. PySpatial streamlines the conventional pipeline by directly operating on computational regions of interest, reducing redundant processing steps. Utilizing rtree-based spatial indexing and matrix-based computation, PySpatial efficiently maps and processes computational regions, significantly accelerating feature extraction while maintaining high accuracy. Our experiments on two datasets-Perivascular Epithelioid Cell (PEC) and data from the Kidney Precision Medicine Project (KPMP)-demonstrate substantial performance improvements. For smaller and sparse objects in PEC datasets, PySpatial achieves nearly a 10-fold speedup compared to standard CellProfiler pipelines. For larger objects, such as glomeruli and arteries in KPMP datasets, PySpatial achieves a 2-fold speedup. These results highlight PySpatial's potential to handle large-scale WSI analysis with enhanced efficiency and accuracy, paving the way for broader applications in digital pathology.
ASIGN: An Anatomy-aware Spatial Imputation Graphic Network for 3D Spatial Transcriptomics
Dec 04, 2024



Spatial transcriptomics (ST) is an emerging technology that enables medical computer vision scientists to automatically interpret the molecular profiles underlying morphological features. Currently, however, most deep learning-based ST analyses are limited to two-dimensional (2D) sections, which can introduce diagnostic errors due to the heterogeneity of pathological tissues across 3D sections. Expanding ST to three-dimensional (3D) volumes is challenging due to the prohibitive costs; a 2D ST acquisition already costs over 50 times more than whole slide imaging (WSI), and a full 3D volume with 10 sections can be an order of magnitude more expensive. To reduce costs, scientists have attempted to predict ST data directly from WSI without performing actual ST acquisition. However, these methods typically yield unsatisfying results. To address this, we introduce a novel problem setting: 3D ST imputation using 3D WSI histology sections combined with a single 2D ST slide. To do so, we present the Anatomy-aware Spatial Imputation Graph Network (ASIGN) for more precise, yet affordable, 3D ST modeling. The ASIGN architecture extends existing 2D spatial relationships into 3D by leveraging cross-layer overlap and similarity-based expansion. Moreover, a multi-level spatial attention graph network integrates features comprehensively across different data sources. We evaluated ASIGN on three public spatial transcriptomics datasets, with experimental results demonstrating that ASIGN achieves state-of-the-art performance on both 2D and 3D scenarios. Code is available at https://github.com/hrlblab/ASIGN.
GloFinder: AI-empowered QuPath Plugin for WSI-level Glomerular Detection, Visualization, and Curation
Nov 27, 2024


Artificial intelligence (AI) has demonstrated significant success in automating the detection of glomeruli, the key functional units of the kidney, from whole slide images (WSIs) in kidney pathology. However, existing open-source tools are often distributed as source code or Docker containers, requiring advanced programming skills that hinder accessibility for non-programmers, such as clinicians. Additionally, current models are typically trained on a single dataset and lack flexibility in adjusting confidence levels for predictions. To overcome these challenges, we introduce GloFinder, a QuPath plugin designed for single-click automated glomeruli detection across entire WSIs with online editing through the graphical user interface (GUI). GloFinder employs CircleNet, an anchor-free detection framework utilizing circle representations for precise object localization, with models trained on approximately 160,000 manually annotated glomeruli. To further enhance accuracy, the plugin incorporates Weighted Circle Fusion (WCF), an ensemble method that combines confidence scores from multiple CircleNet models to produce refined predictions, achieving superior performance in glomerular detection. GloFinder enables direct visualization and editing of results in QuPath, facilitating seamless interaction for clinicians and providing a powerful tool for nephropathology research and clinical practice.