 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeOn the Connection Between MPNN and Graph Transformer
Feb 03, 2023Graph Transformer (GT) recently has emerged as a new paradigm of graph learning algorithms, outperforming the previously popular Message Passing Neural Network (MPNN) on multiple benchmarks. Previous work (Kim et al., 2022) shows that with proper position embedding, GT can approximate MPNN arbitrarily well, implying that GT is at least as powerful as MPNN. In this paper, we study the inverse connection and show that MPNN with virtual node (VN), a commonly used heuristic with little theoretical understanding, is powerful enough to arbitrarily approximate the self-attention layer of GT. In particular, we first show that if we consider one type of linear transformer, the so-called Performer/Linear Transformer (Choromanski et al., 2020; Katharopoulos et al., 2020), then MPNN + VN with only O(1) depth and O(1) width can approximate a self-attention layer in Performer/Linear Transformer. Next, via a connection between MPNN + VN and DeepSets, we prove the MPNN + VN with O(n^d) width and O(1) depth can approximate the self-attention layer arbitrarily well, where d is the input feature dimension. Lastly, under some assumptions, we provide an explicit construction of MPNN + VN with O(1) width and O(n) depth approximating the self-attention layer in GT arbitrarily well. On the empirical side, we demonstrate that 1) MPNN + VN is a surprisingly strong baseline, outperforming GT on the recently proposed Long Range Graph Benchmark (LRGB) dataset, 2) our MPNN + VN improves over early implementation on a wide range of OGB datasets and 3) MPNN + VN outperforms Linear Transformer and MPNN on the climate modeling task.
Copula Conformal Prediction for Multi-step Time Series Forecasting
Dec 06, 2022Accurate uncertainty measurement is a key step to building robust and reliable machine learning systems. Conformal prediction is a distribution-free uncertainty quantification algorithm popular for its ease of implementation, statistical coverage guarantees, and versatility for underlying forecasters. However, existing conformal prediction algorithms for time series are limited to single-step prediction without considering the temporal dependency. In this paper we propose a Copula Conformal Prediction algorithm for multivariate, multi-step Time Series forecasting, CopulaCPTS. On several synthetic and real-world multivariate time series datasets, we show that CopulaCPTS produces more calibrated and sharp confidence intervals for multi-step prediction tasks than existing techniques.
Symmetries, flat minima, and the conserved quantities of gradient flow
Oct 31, 2022



Empirical studies of the loss landscape of deep networks have revealed that many local minima are connected through low-loss valleys. Ensemble models sampling different parts of a low-loss valley have reached SOTA performance. Yet, little is known about the theoretical origin of such valleys. We present a general framework for finding continuous symmetries in the parameter space, which carve out low-loss valleys. Importantly, we introduce a novel set of nonlinear, data-dependent symmetries for neural networks. These symmetries can transform a trained model such that it performs similarly on new samples. We then show that conserved quantities associated with linear symmetries can be used to define coordinates along low-loss valleys. The conserved quantities help reveal that using common initialization methods, gradient flow only explores a small part of the global minimum. By relating conserved quantities to convergence rate and sharpness of the minimum, we provide insights on how initialization impacts convergence and generalizability. We also find the nonlinear action to be viable for ensemble building to improve robustness under certain adversarial attacks.
Koopman Neural Forecaster for Time Series with Temporal Distribution Shifts
Oct 10, 2022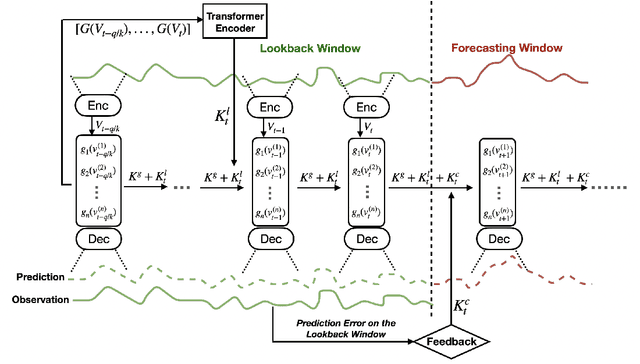
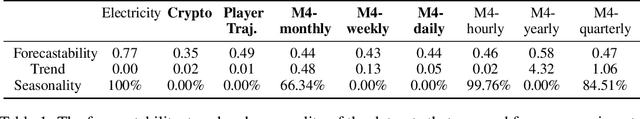
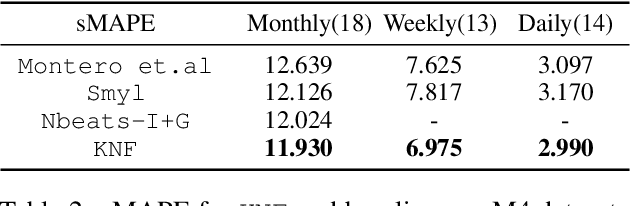
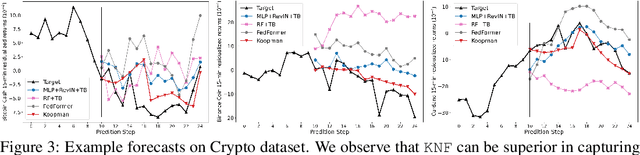
Temporal distributional shifts, with underlying dynamics changing over time, frequently occur in real-world time series, and pose a fundamental challenge for deep neural networks (DNNs). In this paper, we propose a novel deep sequence model based on the Koopman theory for time series forecasting: Koopman Neural Forecaster (KNF) that leverages DNNs to learn the linear Koopman space and the coefficients of chosen measurement functions. KNF imposes appropriate inductive biases for improved robustness against distributional shifts, employing both a global operator to learn shared characteristics, and a local operator to capture changing dynamics, as well as a specially-designed feedback loop to continuously update the learnt operators over time for rapidly varying behaviors. To the best of our knowledge, this is the first time that Koopman theory is applied to real-world chaotic time series without known governing laws. We demonstrate that KNF achieves the superior performance compared to the alternatives, on multiple time series datasets that are shown to suffer from distribution shifts.
Predicting the Future of AI with AI: High-quality link prediction in an exponentially growing knowledge network
Sep 23, 2022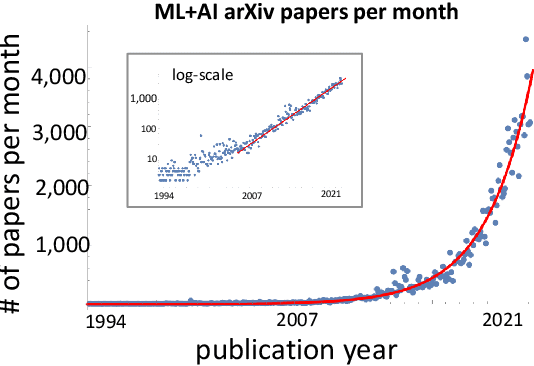
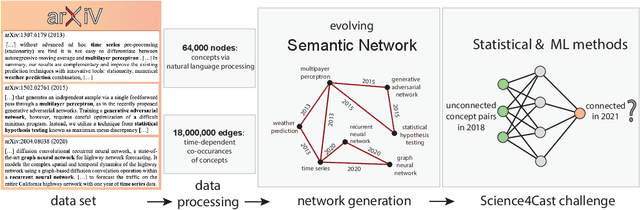
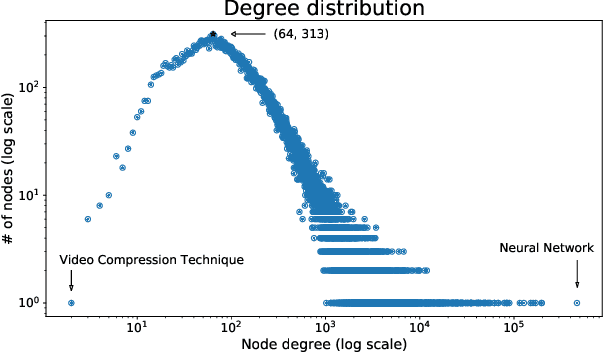
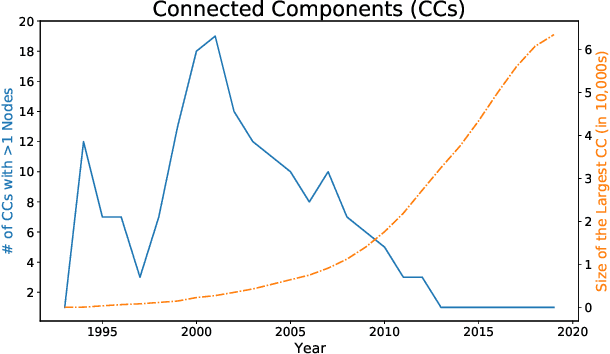
A tool that could suggest new personalized research directions and ideas by taking insights from the scientific literature could significantly accelerate the progress of science. A field that might benefit from such an approach is artificial intelligence (AI) research, where the number of scientific publications has been growing exponentially over the last years, making it challenging for human researchers to keep track of the progress. Here, we use AI techniques to predict the future research directions of AI itself. We develop a new graph-based benchmark based on real-world data -- the Science4Cast benchmark, which aims to predict the future state of an evolving semantic network of AI. For that, we use more than 100,000 research papers and build up a knowledge network with more than 64,000 concept nodes. We then present ten diverse methods to tackle this task, ranging from pure statistical to pure learning methods. Surprisingly, the most powerful methods use a carefully curated set of network features, rather than an end-to-end AI approach. It indicates a great potential that can be unleashed for purely ML approaches without human knowledge. Ultimately, better predictions of new future research directions will be a crucial component of more advanced research suggestion tools.
Data Augmentation vs. Equivariant Networks: A Theory of Generalization on Dynamics Forecasting
Jun 19, 2022Exploiting symmetry in dynamical systems is a powerful way to improve the generalization of deep learning. The model learns to be invariant to transformation and hence is more robust to distribution shift. Data augmentation and equivariant networks are two major approaches to injecting symmetry into learning. However, their exact role in improving generalization is not well understood. In this work, we derive the generalization bounds for data augmentation and equivariant networks, characterizing their effect on learning in a unified framework. Unlike most prior theories for the i.i.d. setting, we focus on non-stationary dynamics forecasting with complex temporal dependencies.
LIMO: Latent Inceptionism for Targeted Molecule Generation
Jun 17, 2022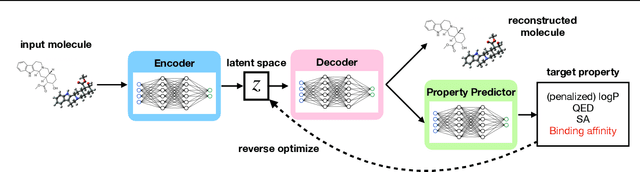
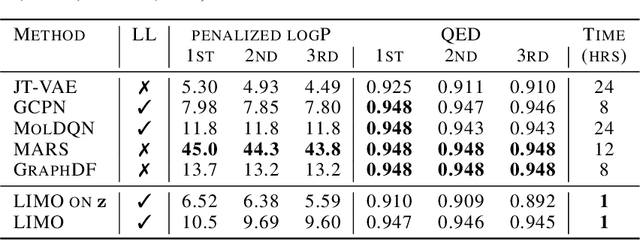
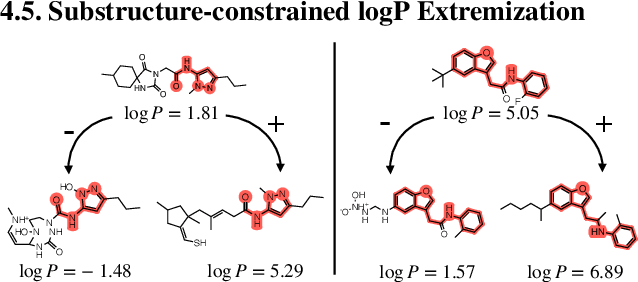

Generation of drug-like molecules with high binding affinity to target proteins remains a difficult and resource-intensive task in drug discovery. Existing approaches primarily employ reinforcement learning, Markov sampling, or deep generative models guided by Gaussian processes, which can be prohibitively slow when generating molecules with high binding affinity calculated by computationally-expensive physics-based methods. We present Latent Inceptionism on Molecules (LIMO), which significantly accelerates molecule generation with an inceptionism-like technique. LIMO employs a variational autoencoder-generated latent space and property prediction by two neural networks in sequence to enable faster gradient-based reverse-optimization of molecular properties. Comprehensive experiments show that LIMO performs competitively on benchmark tasks and markedly outperforms state-of-the-art techniques on the novel task of generating drug-like compounds with high binding affinity, reaching nanomolar range against two protein targets. We corroborate these docking-based results with more accurate molecular dynamics-based calculations of absolute binding free energy and show that one of our generated drug-like compounds has a predicted $K_D$ (a measure of binding affinity) of $6 \cdot 10^{-14}$ M against the human estrogen receptor, well beyond the affinities of typical early-stage drug candidates and most FDA-approved drugs to their respective targets. Code is available at https://github.com/Rose-STL-Lab/LIMO.
Multi-fidelity Hierarchical Neural Processes
Jun 10, 2022

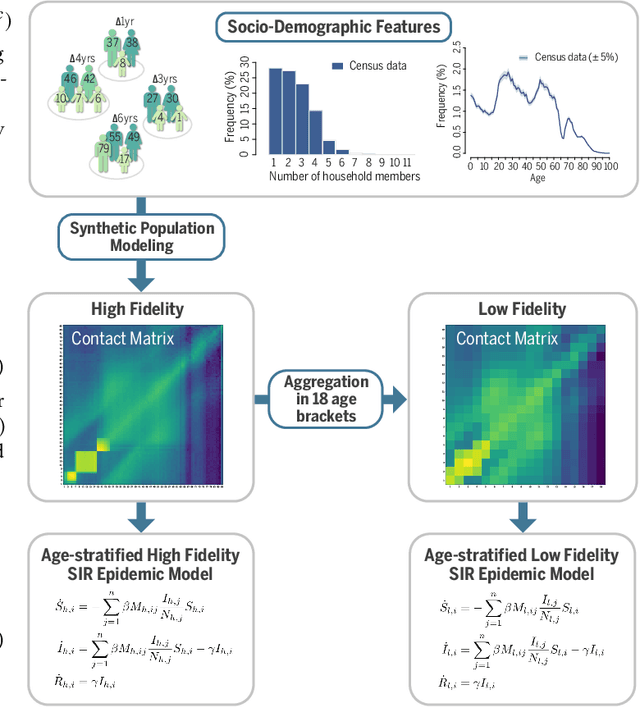
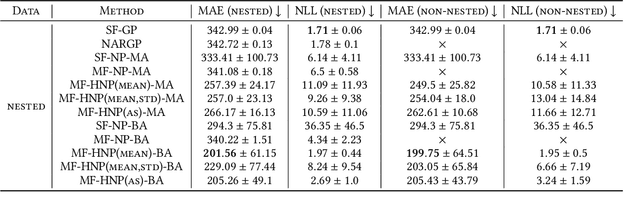
Science and engineering fields use computer simulation extensively. These simulations are often run at multiple levels of sophistication to balance accuracy and efficiency. Multi-fidelity surrogate modeling reduces the computational cost by fusing different simulation outputs. Cheap data generated from low-fidelity simulators can be combined with limited high-quality data generated by an expensive high-fidelity simulator. Existing methods based on Gaussian processes rely on strong assumptions of the kernel functions and can hardly scale to high-dimensional settings. We propose Multi-fidelity Hierarchical Neural Processes (MF-HNP), a unified neural latent variable model for multi-fidelity surrogate modeling. MF-HNP inherits the flexibility and scalability of Neural Processes. The latent variables transform the correlations among different fidelity levels from observations to latent space. The predictions across fidelities are conditionally independent given the latent states. It helps alleviate the error propagation issue in existing methods. MF-HNP is flexible enough to handle non-nested high dimensional data at different fidelity levels with varying input and output dimensions. We evaluate MF-HNP on epidemiology and climate modeling tasks, achieving competitive performance in terms of accuracy and uncertainty estimation. In contrast to deep Gaussian Processes with only low-dimensional (< 10) tasks, our method shows great promise for speeding up high-dimensional complex simulations (over 7000 for epidemiology modeling and 45000 for climate modeling).
Faster Optimization on Sparse Graphs via Neural Reparametrization
May 26, 2022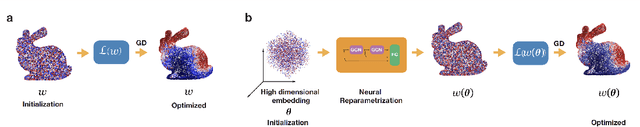
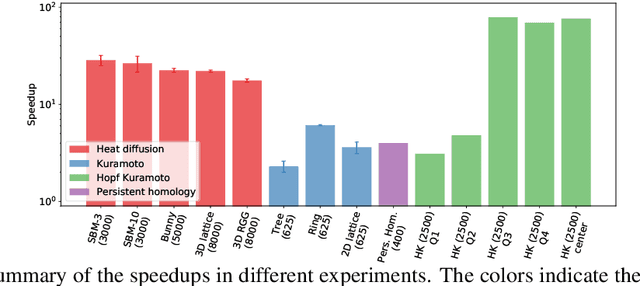
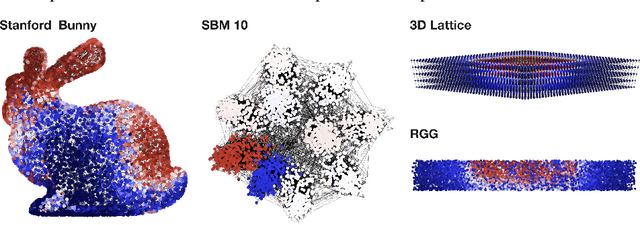
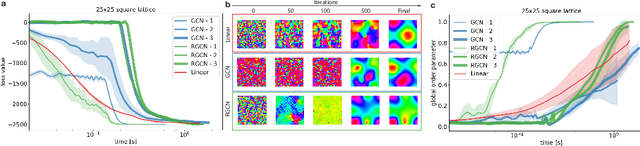
In mathematical optimization, second-order Newton's methods generally converge faster than first-order methods, but they require the inverse of the Hessian, hence are computationally expensive. However, we discover that on sparse graphs, graph neural networks (GNN) can implement an efficient Quasi-Newton method that can speed up optimization by a factor of 10-100x. Our method, neural reparametrization, modifies the optimization parameters as the output of a GNN to reshape the optimization landscape. Using a precomputed Hessian as the propagation rule, the GNN can effectively utilize the second-order information, reaching a similar effect as adaptive gradient methods. As our method solves optimization through architecture design, it can be used in conjunction with any optimizers such as Adam and RMSProp. We show the application of our method on scientifically relevant problems including heat diffusion, synchronization and persistent homology.
Symmetry Teleportation for Accelerated Optimization
May 21, 2022
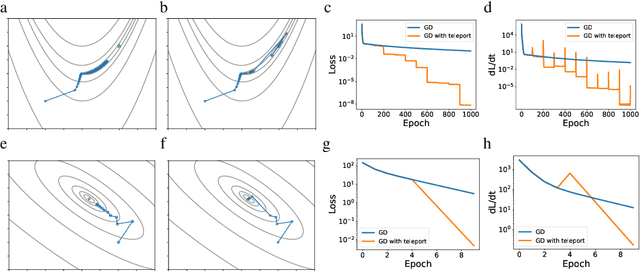
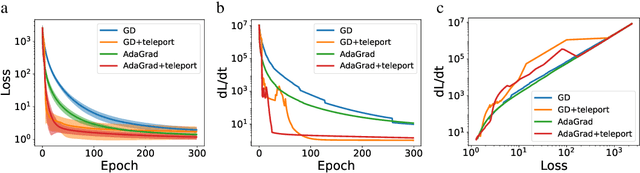
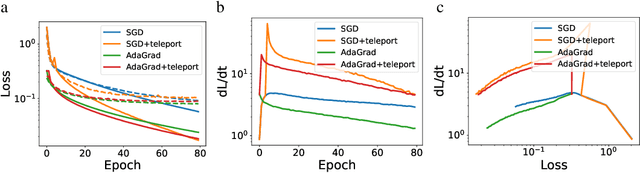
Existing gradient-based optimization methods update the parameters locally, in a direction that minimizes the loss function. We study a different approach, symmetry teleportation, that allows the parameters to travel a large distance on the loss level set, in order to improve the convergence speed in subsequent steps. Teleportation exploits parameter space symmetries of the optimization problem and transforms parameters while keeping the loss invariant. We derive the loss-invariant group actions for test functions and multi-layer neural networks, and prove a necessary condition of when teleportation improves convergence rate. We also show that our algorithm is closely related to second order methods. Experimentally, we show that teleportation improves the convergence speed of gradient descent and AdaGrad for several optimization problems including test functions, multi-layer regressions, and MNIST classification.