 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAutomatic Integration for Spatiotemporal Neural Point Processes
Oct 09, 2023Learning continuous-time point processes is essential to many discrete event forecasting tasks. However, integration poses a major challenge, particularly for spatiotemporal point processes (STPPs), as it involves calculating the likelihood through triple integrals over space and time. Existing methods for integrating STPP either assume a parametric form of the intensity function, which lacks flexibility; or approximating the intensity with Monte Carlo sampling, which introduces numerical errors. Recent work by Omi et al. [2019] proposes a dual network or AutoInt approach for efficient integration of flexible intensity function. However, the method only focuses on the 1D temporal point process. In this paper, we introduce a novel paradigm: AutoSTPP (Automatic Integration for Spatiotemporal Neural Point Processes) that extends the AutoInt approach to 3D STPP. We show that direct extension of the previous work overly constrains the intensity function, leading to poor performance. We prove consistency of AutoSTPP and validate it on synthetic data and benchmark real world datasets, showcasing its significant advantage in recovering complex intensity functions from irregular spatiotemporal events, particularly when the intensity is sharply localized.
Latent Space Symmetry Discovery
Sep 29, 2023



Equivariant neural networks require explicit knowledge of the symmetry group. Automatic symmetry discovery methods aim to relax this constraint and learn invariance and equivariance from data. However, existing symmetry discovery methods are limited to linear symmetries in their search space and cannot handle the complexity of symmetries in real-world, often high-dimensional data. We propose a novel generative model, Latent LieGAN (LaLiGAN), which can discover nonlinear symmetries from data. It learns a mapping from data to a latent space where the symmetries become linear and simultaneously discovers symmetries in the latent space. Theoretically, we show that our method can express any nonlinear symmetry under certain conditions. Experimentally, our method can capture the intrinsic symmetry in high-dimensional observations, which results in a well-structured latent space that is useful for other downstream tasks. We demonstrate the use cases for LaLiGAN in improving equation discovery and long-term forecasting for various dynamical systems.
Artificial Intelligence for Science in Quantum, Atomistic, and Continuum Systems
Jul 17, 2023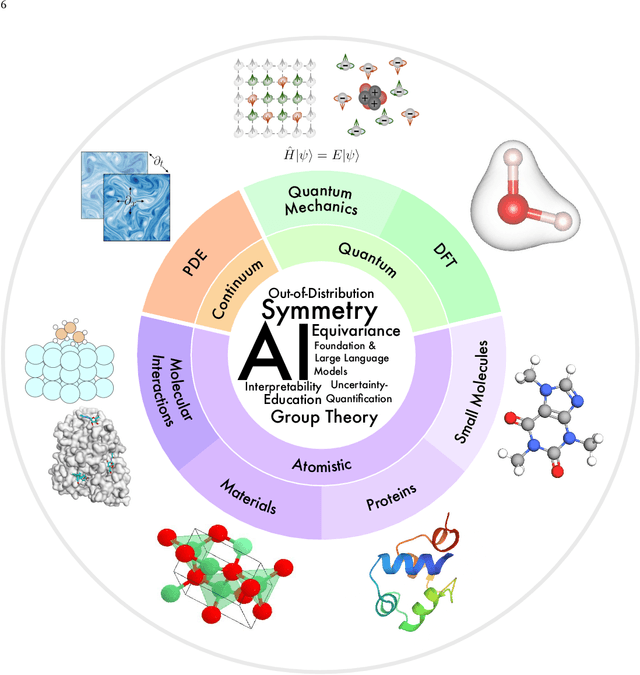



Advances in artificial intelligence (AI) are fueling a new paradigm of discoveries in natural sciences. Today, AI has started to advance natural sciences by improving, accelerating, and enabling our understanding of natural phenomena at a wide range of spatial and temporal scales, giving rise to a new area of research known as AI for science (AI4Science). Being an emerging research paradigm, AI4Science is unique in that it is an enormous and highly interdisciplinary area. Thus, a unified and technical treatment of this field is needed yet challenging. This paper aims to provide a technically thorough account of a subarea of AI4Science; namely, AI for quantum, atomistic, and continuum systems. These areas aim at understanding the physical world from the subatomic (wavefunctions and electron density), atomic (molecules, proteins, materials, and interactions), to macro (fluids, climate, and subsurface) scales and form an important subarea of AI4Science. A unique advantage of focusing on these areas is that they largely share a common set of challenges, thereby allowing a unified and foundational treatment. A key common challenge is how to capture physics first principles, especially symmetries, in natural systems by deep learning methods. We provide an in-depth yet intuitive account of techniques to achieve equivariance to symmetry transformations. We also discuss other common technical challenges, including explainability, out-of-distribution generalization, knowledge transfer with foundation and large language models, and uncertainty quantification. To facilitate learning and education, we provide categorized lists of resources that we found to be useful. We strive to be thorough and unified and hope this initial effort may trigger more community interests and efforts to further advance AI4Science.
ClimSim: An open large-scale dataset for training high-resolution physics emulators in hybrid multi-scale climate simulators
Jun 16, 2023Modern climate projections lack adequate spatial and temporal resolution due to computational constraints. A consequence is inaccurate and imprecise prediction of critical processes such as storms. Hybrid methods that combine physics with machine learning (ML) have introduced a new generation of higher fidelity climate simulators that can sidestep Moore's Law by outsourcing compute-hungry, short, high-resolution simulations to ML emulators. However, this hybrid ML-physics simulation approach requires domain-specific treatment and has been inaccessible to ML experts because of lack of training data and relevant, easy-to-use workflows. We present ClimSim, the largest-ever dataset designed for hybrid ML-physics research. It comprises multi-scale climate simulations, developed by a consortium of climate scientists and ML researchers. It consists of 5.7 billion pairs of multivariate input and output vectors that isolate the influence of locally-nested, high-resolution, high-fidelity physics on a host climate simulator's macro-scale physical state. The dataset is global in coverage, spans multiple years at high sampling frequency, and is designed such that resulting emulators are compatible with downstream coupling into operational climate simulators. We implement a range of deterministic and stochastic regression baselines to highlight the ML challenges and their scoring. The data (https://huggingface.co/datasets/LEAP/ClimSim_high-res) and code (https://leap-stc.github.io/ClimSim) are released openly to support the development of hybrid ML-physics and high-fidelity climate simulations for the benefit of science and society.
DYffusion: A Dynamics-informed Diffusion Model for Spatiotemporal Forecasting
Jun 03, 2023While diffusion models can successfully generate data and make predictions, they are predominantly designed for static images. We propose an approach for training diffusion models for dynamics forecasting that leverages the temporal dynamics encoded in the data, directly coupling it with the diffusion steps in the network. We train a stochastic, time-conditioned interpolator and a backbone forecaster network that mimic the forward and reverse processes of conventional diffusion models, respectively. This design choice naturally encodes multi-step and long-range forecasting capabilities, allowing for highly flexible, continuous-time sampling trajectories and the ability to trade-off performance with accelerated sampling at inference time. In addition, the dynamics-informed diffusion process imposes a strong inductive bias, allowing for improved computational efficiency compared to traditional Gaussian noise-based diffusion models. Our approach performs competitively on probabilistic skill score metrics in complex dynamics forecasting of sea surface temperatures, Navier-Stokes flows, and spring mesh systems.
Improving Convergence and Generalization Using Parameter Symmetries
May 22, 2023



In overparametrized models, different values of the parameters may result in the same loss value. Parameter space symmetries are transformations that change the model parameters but leave the loss invariant. Teleportation applies such transformations to accelerate optimization. However, the exact mechanism behind this algorithm's success is not well understood. In this paper, we show that teleportation not only speeds up optimization in the short-term, but gives overall faster time to convergence. Additionally, we show that teleporting to minima with different curvatures improves generalization and provide insights on the connection between the curvature of the minima and generalization ability. Finally, we show that integrating teleportation into a wide range of optimization algorithms and optimization-based meta-learning improves convergence.
Disentangled Multi-Fidelity Deep Bayesian Active Learning
May 07, 2023To balance quality and cost, various domain areas of science and engineering run simulations at multiple levels of sophistication. Multi-fidelity active learning aims to learn a direct mapping from input parameters to simulation outputs by actively acquiring data from multiple fidelity levels. However, existing approaches based on Gaussian processes are hardly scalable to high-dimensional data. Other deep learning-based methods use the hierarchical structure, which only supports passing information from low-fidelity to high-fidelity. This approach also leads to the undesirable propagation of errors from low-fidelity representations to high-fidelity ones. We propose a novel disentangled deep Bayesian learning framework for multi-fidelity active learning, that learns the surrogate models conditioned on the distribution of functions at multiple fidelities.
Long-term Forecasting with TiDE: Time-series Dense Encoder
Apr 27, 2023



Recent work has shown that simple linear models can outperform several Transformer based approaches in long term time-series forecasting. Motivated by this, we propose a Multi-layer Perceptron (MLP) based encoder-decoder model, Time-series Dense Encoder (TiDE), for long-term time-series forecasting that enjoys the simplicity and speed of linear models while also being able to handle covariates and non-linear dependencies. Theoretically, we prove that the simplest linear analogue of our model can achieve near optimal error rate for linear dynamical systems (LDS) under some assumptions. Empirically, we show that our method can match or outperform prior approaches on popular long-term time-series forecasting benchmarks while being 5-10x faster than the best Transformer based model.
Understanding why shooters shoot -- An AI-powered engine for basketball performance profiling
Mar 17, 2023



Understanding player shooting profiles is an essential part of basketball analysis: knowing where certain opposing players like to shoot from can help coaches neutralize offensive gameplans from their opponents; understanding where their players are most comfortable can lead them to developing more effective offensive strategies. An automatic tool that can provide these performance profiles in a timely manner can become invaluable for coaches to maximize both the effectiveness of their game plan as well as the time dedicated to practice and other related activities. Additionally, basketball is dictated by many variables, such as playstyle and game dynamics, that can change the flow of the game and, by extension, player performance profiles. It is crucial that the performance profiles can reflect the diverse playstyles, as well as the fast-changing dynamics of the game. We present a tool that can visualize player performance profiles in a timely manner while taking into account factors such as play-style and game dynamics. Our approach generates interpretable heatmaps that allow us to identify and analyze how non-spatial factors, such as game dynamics or playstyle, affect player performance profiles.
Generative Adversarial Symmetry Discovery
Feb 08, 2023Despite the success of equivariant neural networks in scientific applications, they require knowing the symmetry group a priori. However, it may be difficult to know the right symmetry to use as an inductive bias in practice and enforcing the wrong symmetry could hurt the performance. In this paper, we propose a framework, LieGAN, to automatically discover equivariances from a dataset using a paradigm akin to generative adversarial training. Specifically, a generator learns a group of transformations applied to the data, which preserves the original distribution and fools the discriminator. LieGAN represents symmetry as interpretable Lie algebra basis and can discover various symmetries such as rotation group $\mathrm{SO}(n)$ and restricted Lorentz group $\mathrm{SO}(1,3)^+$ in trajectory prediction and top quark tagging tasks. The learned symmetry can also be readily used in several existing equivariant neural networks to improve accuracy and generalization in prediction.