 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEquiBind: Geometric Deep Learning for Drug Binding Structure Prediction
Feb 07, 2022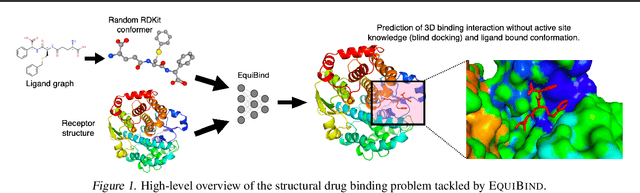
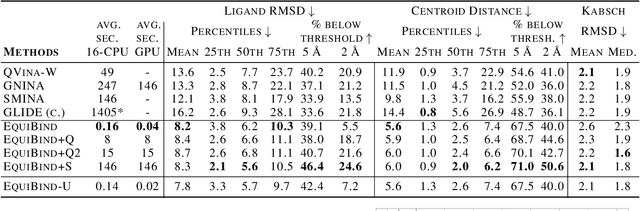
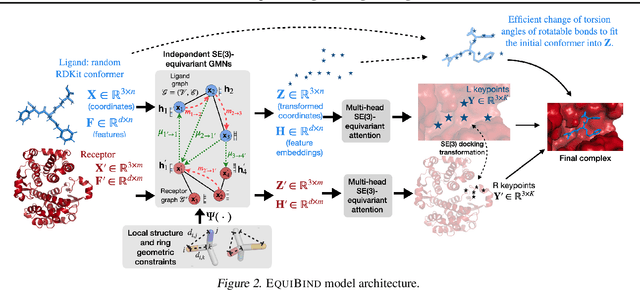
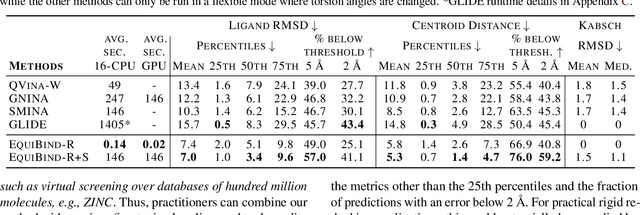
Predicting how a drug-like molecule binds to a specific protein target is a core problem in drug discovery. An extremely fast computational binding method would enable key applications such as fast virtual screening or drug engineering. Existing methods are computationally expensive as they rely on heavy candidate sampling coupled with scoring, ranking, and fine-tuning steps. We challenge this paradigm with EquiBind, an SE(3)-equivariant geometric deep learning model performing direct-shot prediction of both i) the receptor binding location (blind docking) and ii) the ligand's bound pose and orientation. EquiBind achieves significant speed-ups and better quality compared to traditional and recent baselines. Further, we show extra improvements when coupling it with existing fine-tuning techniques at the cost of increased running time. Finally, we propose a novel and fast fine-tuning model that adjusts torsion angles of a ligand's rotatable bonds based on closed-form global minima of the von Mises angular distance to a given input atomic point cloud, avoiding previous expensive differential evolution strategies for energy minimization.
Syfer: Neural Obfuscation for Private Data Release
Jan 28, 2022



Balancing privacy and predictive utility remains a central challenge for machine learning in healthcare. In this paper, we develop Syfer, a neural obfuscation method to protect against re-identification attacks. Syfer composes trained layers with random neural networks to encode the original data (e.g. X-rays) while maintaining the ability to predict diagnoses from the encoded data. The randomness in the encoder acts as the private key for the data owner. We quantify privacy as the number of attacker guesses required to re-identify a single image (guesswork). We propose a contrastive learning algorithm to estimate guesswork. We show empirically that differentially private methods, such as DP-Image, obtain privacy at a significant loss of utility. In contrast, Syfer achieves strong privacy while preserving utility. For example, X-ray classifiers built with DP-image, Syfer, and original data achieve average AUCs of 0.53, 0.78, and 0.86, respectively.
Independent SE-Equivariant Models for End-to-End Rigid Protein Docking
Nov 15, 2021



Protein complex formation is a central problem in biology, being involved in most of the cell's processes, and essential for applications, e.g. drug design or protein engineering. We tackle rigid body protein-protein docking, i.e., computationally predicting the 3D structure of a protein-protein complex from the individual unbound structures, assuming no conformational change within the proteins happens during binding. We design a novel pairwise-independent SE(3)-equivariant graph matching network to predict the rotation and translation to place one of the proteins at the right docked position relative to the second protein. We mathematically guarantee a basic principle: the predicted complex is always identical regardless of the initial locations and orientations of the two structures. Our model, named EquiDock, approximates the binding pockets and predicts the docking poses using keypoint matching and alignment, achieved through optimal transport and a differentiable Kabsch algorithm. Empirically, we achieve significant running time improvements and often outperform existing docking software despite not relying on heavy candidate sampling, structure refinement, or templates.
Fragment-based Sequential Translation for Molecular Optimization
Oct 26, 2021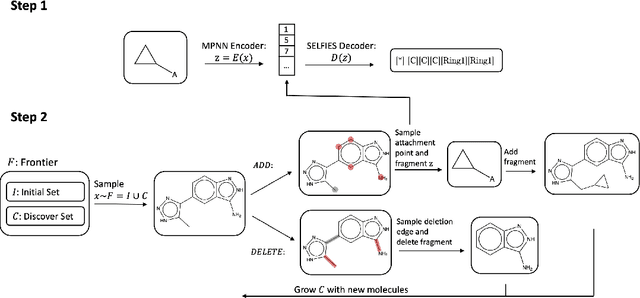
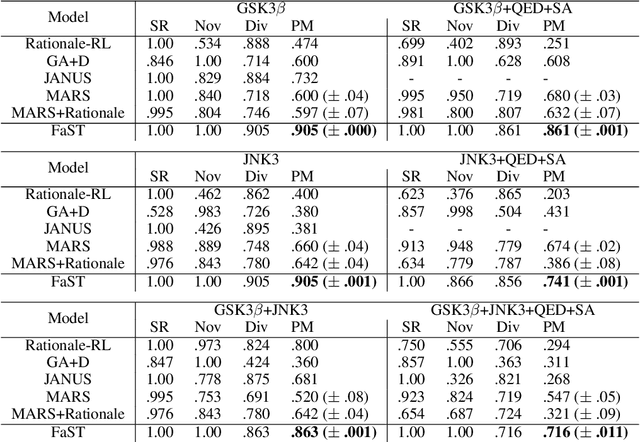
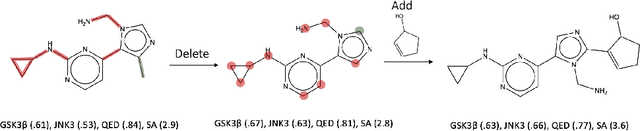
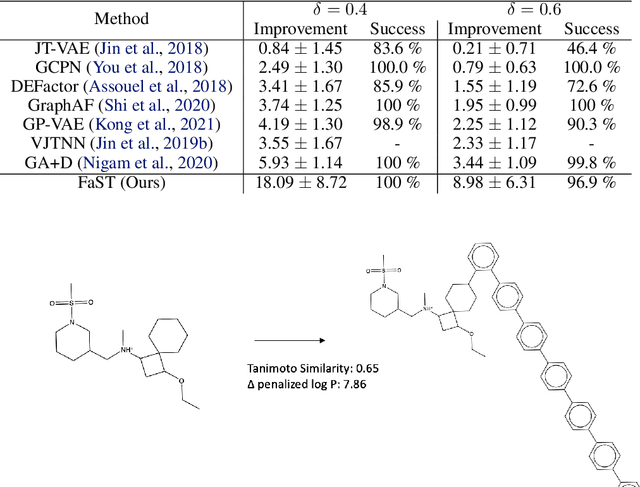
Searching for novel molecular compounds with desired properties is an important problem in drug discovery. Many existing frameworks generate molecules one atom at a time. We instead propose a flexible editing paradigm that generates molecules using learned molecular fragments--meaningful substructures of molecules. To do so, we train a variational autoencoder (VAE) to encode molecular fragments in a coherent latent space, which we then utilize as a vocabulary for editing molecules to explore the complex chemical property space. Equipped with the learned fragment vocabulary, we propose Fragment-based Sequential Translation (FaST), which learns a reinforcement learning (RL) policy to iteratively translate model-discovered molecules into increasingly novel molecules while satisfying desired properties. Empirical evaluation shows that FaST significantly improves over state-of-the-art methods on benchmark single/multi-objective molecular optimization tasks.
Iterative Refinement Graph Neural Network for Antibody Sequence-Structure Co-design
Oct 15, 2021
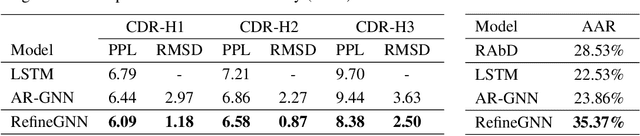
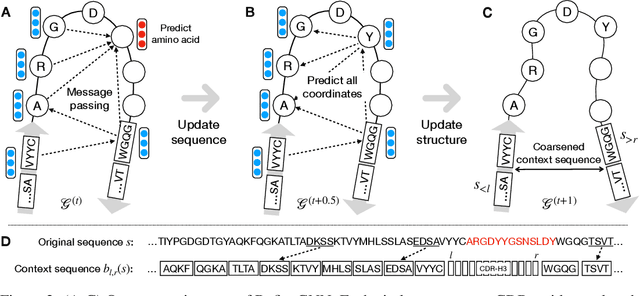
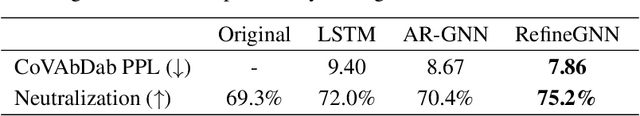
Antibodies are versatile proteins that bind to pathogens like viruses and stimulate the adaptive immune system. The specificity of antibody binding is determined by complementarity-determining regions (CDRs) at the tips of these Y-shaped proteins. In this paper, we propose a generative model to automatically design the CDRs of antibodies with enhanced binding specificity or neutralization capabilities. Previous generative approaches formulate protein design as a structure-conditioned sequence generation task, assuming the desired 3D structure is given a priori. In contrast, we propose to co-design the sequence and 3D structure of CDRs as graphs. Our model unravels a sequence autoregressively while iteratively refining its predicted global structure. The inferred structure in turn guides subsequent residue choices. For efficiency, we model the conditional dependence between residues inside and outside of a CDR in a coarse-grained manner. Our method achieves superior log-likelihood on the test set and outperforms previous baselines in designing antibodies capable of neutralizing the SARS-CoV-2 virus.
Crystal Diffusion Variational Autoencoder for Periodic Material Generation
Oct 12, 2021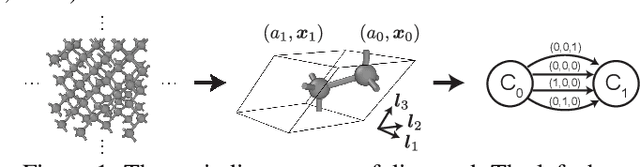

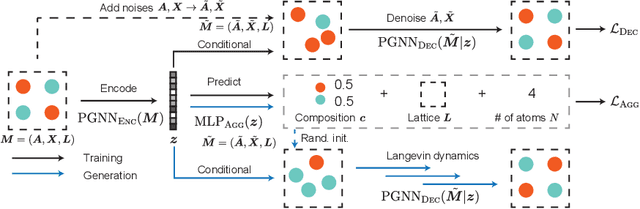
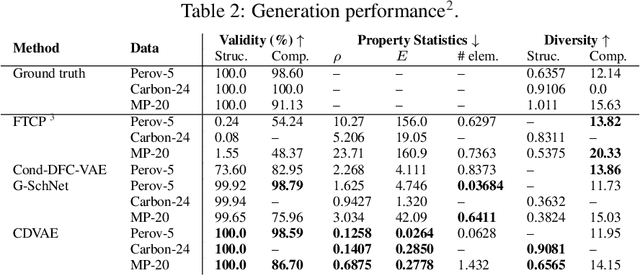
Generating the periodic structure of stable materials is a long-standing challenge for the material design community. This task is difficult because stable materials only exist in a low-dimensional subspace of all possible periodic arrangements of atoms: 1) the coordinates must lie in the local energy minimum defined by quantum mechanics, and 2) global stability also requires the structure to follow the complex, yet specific bonding preferences between different atom types. Existing methods fail to incorporate these factors and often lack proper invariances. We propose a Crystal Diffusion Variational Autoencoder (CDVAE) that captures the physical inductive bias of material stability. By learning from the data distribution of stable materials, the decoder generates materials in a diffusion process that moves atomic coordinates towards a lower energy state and updates atom types to satisfy bonding preferences between neighbors. Our model also explicitly encodes interactions across periodic boundaries and respects permutation, translation, rotation, and periodic invariances. We significantly outperform past methods in three tasks: 1) reconstructing the input structure, 2) generating valid, diverse, and realistic materials, and 3) generating materials that optimize a specific property. We also provide several standard datasets and evaluation metrics for the broader machine learning community.
Learning Stable Classifiers by Transferring Unstable Features
Jun 15, 2021

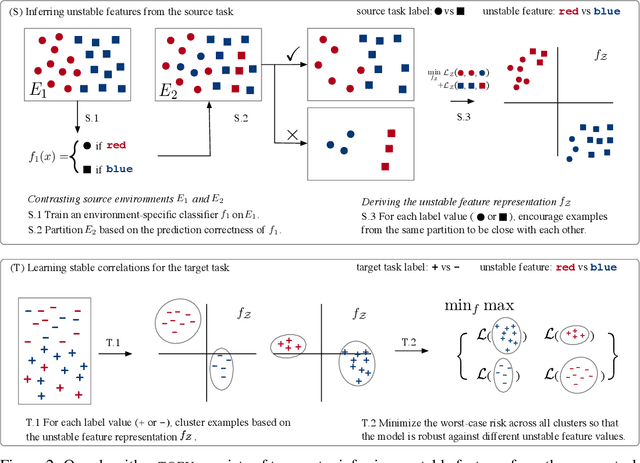
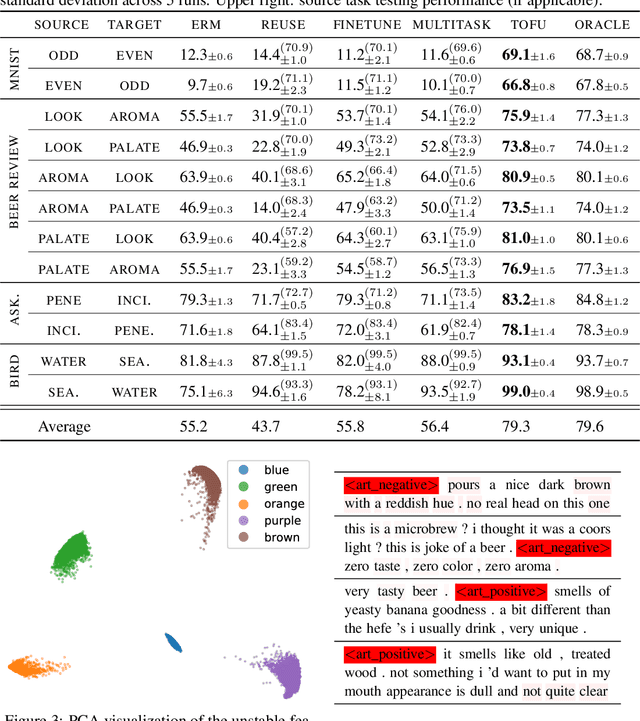
We study transfer learning in the presence of spurious correlations. We experimentally demonstrate that directly transferring the stable feature extractor learned on the source task may not eliminate these biases for the target task. However, we hypothesize that the unstable features in the source task and those in the target task are directly related. By explicitly informing the target classifier of the source task's unstable features, we can regularize the biases in the target task. Specifically, we derive a representation that encodes the unstable features by contrasting different data environments in the source task. On the target task, we cluster data from this representation, and achieve robustness by minimizing the worst-case risk across all clusters. We evaluate our method on both text and image classifications. Empirical results demonstrate that our algorithm is able to maintain robustness on the target task, outperforming the best baseline by 22.9% in absolute accuracy across 12 transfer settings. Our code is available at https://github.com/YujiaBao/Tofu.
GeoMol: Torsional Geometric Generation of Molecular 3D Conformer Ensembles
Jun 08, 2021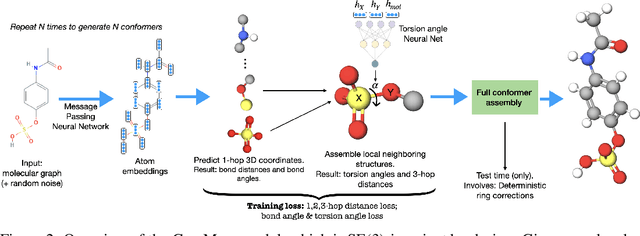
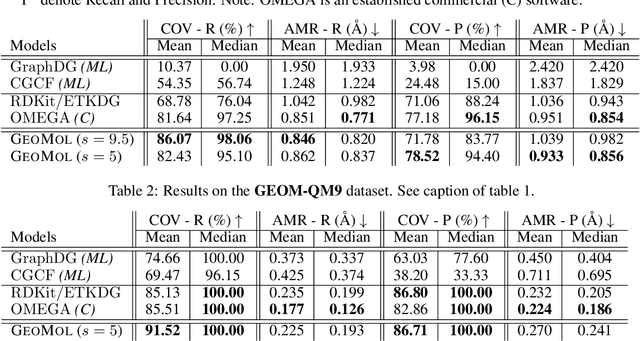
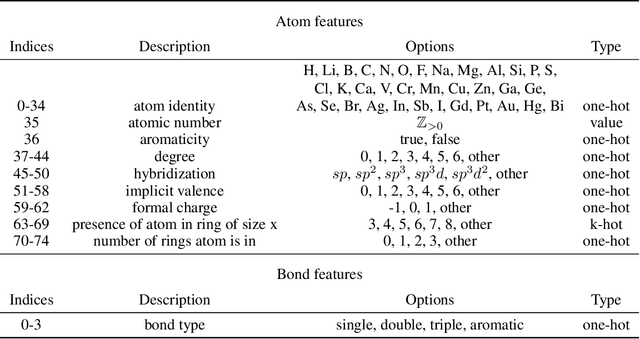
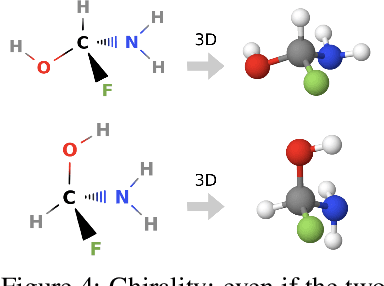
Prediction of a molecule's 3D conformer ensemble from the molecular graph holds a key role in areas of cheminformatics and drug discovery. Existing generative models have several drawbacks including lack of modeling important molecular geometry elements (e.g. torsion angles), separate optimization stages prone to error accumulation, and the need for structure fine-tuning based on approximate classical force-fields or computationally expensive methods such as metadynamics with approximate quantum mechanics calculations at each geometry. We propose GeoMol--an end-to-end, non-autoregressive and SE(3)-invariant machine learning approach to generate distributions of low-energy molecular 3D conformers. Leveraging the power of message passing neural networks (MPNNs) to capture local and global graph information, we predict local atomic 3D structures and torsion angles, avoiding unnecessary over-parameterization of the geometric degrees of freedom (e.g. one angle per non-terminal bond). Such local predictions suffice both for the training loss computation, as well as for the full deterministic conformer assembly (at test time). We devise a non-adversarial optimal transport based loss function to promote diverse conformer generation. GeoMol predominantly outperforms popular open-source, commercial, or state-of-the-art machine learning (ML) models, while achieving significant speed-ups. We expect such differentiable 3D structure generators to significantly impact molecular modeling and related applications.
NeuraCrypt: Hiding Private Health Data via Random Neural Networks for Public Training
Jun 04, 2021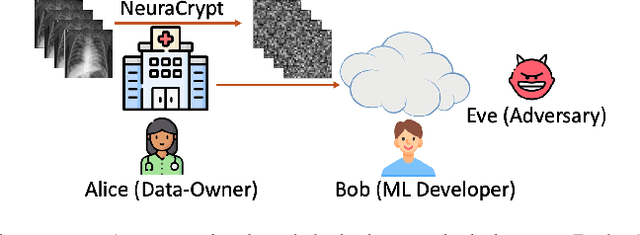
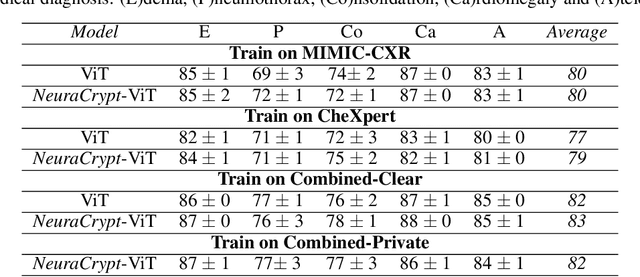
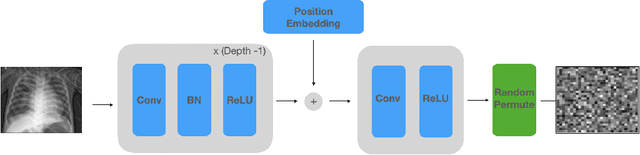

Balancing the needs of data privacy and predictive utility is a central challenge for machine learning in healthcare. In particular, privacy concerns have led to a dearth of public datasets, complicated the construction of multi-hospital cohorts and limited the utilization of external machine learning resources. To remedy this, new methods are required to enable data owners, such as hospitals, to share their datasets publicly, while preserving both patient privacy and modeling utility. We propose NeuraCrypt, a private encoding scheme based on random deep neural networks. NeuraCrypt encodes raw patient data using a randomly constructed neural network known only to the data-owner, and publishes both the encoded data and associated labels publicly. From a theoretical perspective, we demonstrate that sampling from a sufficiently rich family of encoding functions offers a well-defined and meaningful notion of privacy against a computationally unbounded adversary with full knowledge of the underlying data-distribution. We propose to approximate this family of encoding functions through random deep neural networks. Empirically, we demonstrate the robustness of our encoding to a suite of adversarial attacks and show that NeuraCrypt achieves competitive accuracy to non-private baselines on a variety of x-ray tasks. Moreover, we demonstrate that multiple hospitals, using independent private encoders, can collaborate to train improved x-ray models. Finally, we release a challenge dataset to encourage the development of new attacks on NeuraCrypt.
Predict then Interpolate: A Simple Algorithm to Learn Stable Classifiers
May 26, 2021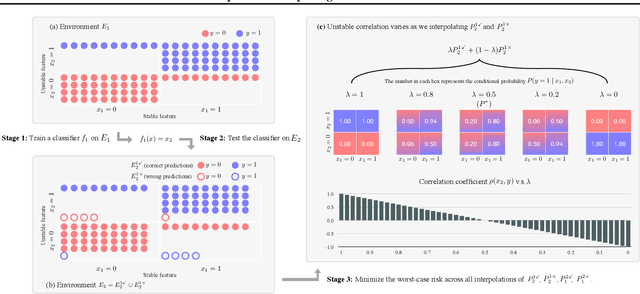
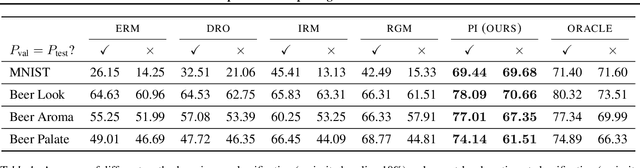
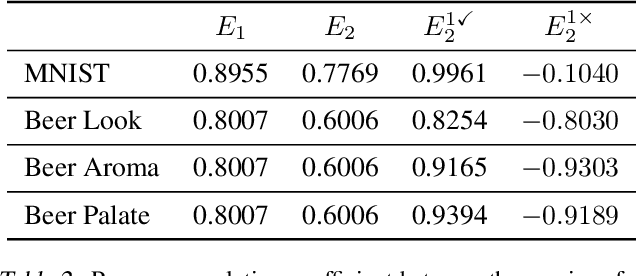
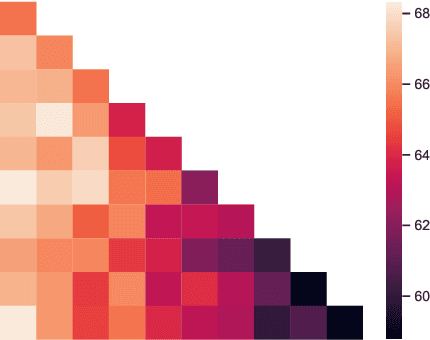
We propose Predict then Interpolate (PI), a simple algorithm for learning correlations that are stable across environments. The algorithm follows from the intuition that when using a classifier trained on one environment to make predictions on examples from another environment, its mistakes are informative as to which correlations are unstable. In this work, we prove that by interpolating the distributions of the correct predictions and the wrong predictions, we can uncover an oracle distribution where the unstable correlation vanishes. Since the oracle interpolation coefficients are not accessible, we use group distributionally robust optimization to minimize the worst-case risk across all such interpolations. We evaluate our method on both text classification and image classification. Empirical results demonstrate that our algorithm is able to learn robust classifiers (outperforms IRM by 23.85% on synthetic environments and 12.41% on natural environments). Our code and data are available at https://github.com/YujiaBao/Predict-then-Interpolate.