 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFast non-autoregressive inverse folding with discrete diffusion
Dec 05, 2023

Generating protein sequences that fold into a intended 3D structure is a fundamental step in de novo protein design. De facto methods utilize autoregressive generation, but this eschews higher order interactions that could be exploited to improve inference speed. We describe a non-autoregressive alternative that performs inference using a constant number of calls resulting in a 23 times speed up without a loss in performance on the CATH benchmark. Conditioned on the 3D structure, we fine-tune ProteinMPNN to perform discrete diffusion with a purity prior over the index sampling order. Our approach gives the flexibility in trading off inference speed and accuracy by modulating the diffusion speed. Code: https://github.com/johnyang101/pmpnndiff
Risk-Controlling Model Selection via Guided Bayesian Optimization
Dec 04, 2023Adjustable hyperparameters of machine learning models typically impact various key trade-offs such as accuracy, fairness, robustness, or inference cost. Our goal in this paper is to find a configuration that adheres to user-specified limits on certain risks while being useful with respect to other conflicting metrics. We solve this by combining Bayesian Optimization (BO) with rigorous risk-controlling procedures, where our core idea is to steer BO towards an efficient testing strategy. Our BO method identifies a set of Pareto optimal configurations residing in a designated region of interest. The resulting candidates are statistically verified and the best-performing configuration is selected with guaranteed risk levels. We demonstrate the effectiveness of our approach on a range of tasks with multiple desiderata, including low error rates, equitable predictions, handling spurious correlations, managing rate and distortion in generative models, and reducing computational costs.
Particle Guidance: non-I.I.D. Diverse Sampling with Diffusion Models
Oct 19, 2023



In light of the widespread success of generative models, a significant amount of research has gone into speeding up their sampling time. However, generative models are often sampled multiple times to obtain a diverse set incurring a cost that is orthogonal to sampling time. We tackle the question of how to improve diversity and sample efficiency by moving beyond the common assumption of independent samples. We propose particle guidance, an extension of diffusion-based generative sampling where a joint-particle time-evolving potential enforces diversity. We analyze theoretically the joint distribution that particle guidance generates, its implications on the choice of potential, and the connections with methods in other disciplines. Empirically, we test the framework both in the setting of conditional image generation, where we are able to increase diversity without affecting quality, and molecular conformer generation, where we reduce the state-of-the-art median error by 13% on average.
Harmonic Self-Conditioned Flow Matching for Multi-Ligand Docking and Binding Site Design
Oct 09, 2023



A significant amount of protein function requires binding small molecules, including enzymatic catalysis. As such, designing binding pockets for small molecules has several impactful applications ranging from drug synthesis to energy storage. Towards this goal, we first develop HarmonicFlow, an improved generative process over 3D protein-ligand binding structures based on our self-conditioned flow matching objective. FlowSite extends this flow model to jointly generate a protein pocket's discrete residue types and the molecule's binding 3D structure. We show that HarmonicFlow improves upon the state-of-the-art generative processes for docking in simplicity, generality, and performance. Enabled by this structure modeling, FlowSite designs binding sites substantially better than baseline approaches and provides the first general solution for binding site design.
Artificial Intelligence for Science in Quantum, Atomistic, and Continuum Systems
Jul 17, 2023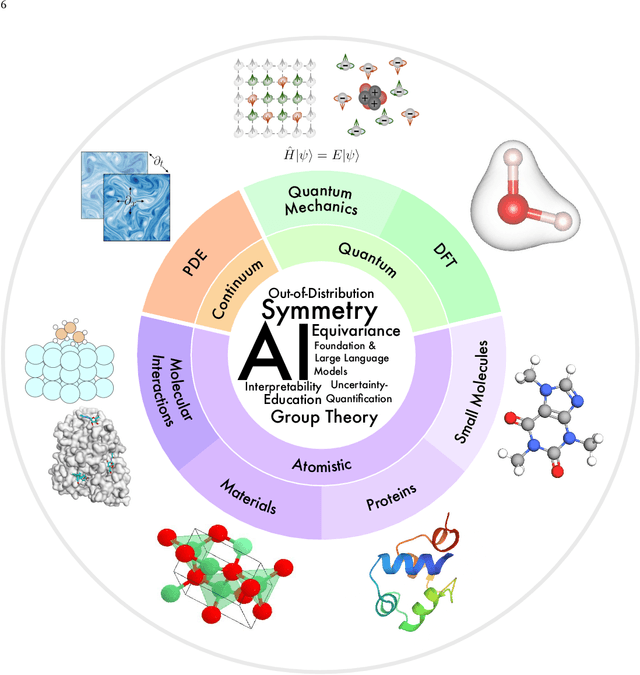



Advances in artificial intelligence (AI) are fueling a new paradigm of discoveries in natural sciences. Today, AI has started to advance natural sciences by improving, accelerating, and enabling our understanding of natural phenomena at a wide range of spatial and temporal scales, giving rise to a new area of research known as AI for science (AI4Science). Being an emerging research paradigm, AI4Science is unique in that it is an enormous and highly interdisciplinary area. Thus, a unified and technical treatment of this field is needed yet challenging. This paper aims to provide a technically thorough account of a subarea of AI4Science; namely, AI for quantum, atomistic, and continuum systems. These areas aim at understanding the physical world from the subatomic (wavefunctions and electron density), atomic (molecules, proteins, materials, and interactions), to macro (fluids, climate, and subsurface) scales and form an important subarea of AI4Science. A unique advantage of focusing on these areas is that they largely share a common set of challenges, thereby allowing a unified and foundational treatment. A key common challenge is how to capture physics first principles, especially symmetries, in natural systems by deep learning methods. We provide an in-depth yet intuitive account of techniques to achieve equivariance to symmetry transformations. We also discuss other common technical challenges, including explainability, out-of-distribution generalization, knowledge transfer with foundation and large language models, and uncertainty quantification. To facilitate learning and education, we provide categorized lists of resources that we found to be useful. We strive to be thorough and unified and hope this initial effort may trigger more community interests and efforts to further advance AI4Science.
Optimizing protein fitness using Gibbs sampling with Graph-based Smoothing
Jul 02, 2023



The ability to design novel proteins with higher fitness on a given task would be revolutionary for many fields of medicine. However, brute-force search through the combinatorially large space of sequences is infeasible. Prior methods constrain search to a small mutational radius from a reference sequence, but such heuristics drastically limit the design space. Our work seeks to remove the restriction on mutational distance while enabling efficient exploration. We propose Gibbs sampling with Graph-based Smoothing (GGS) which iteratively applies Gibbs with gradients to propose advantageous mutations using graph-based smoothing to remove noisy gradients that lead to false positives. Our method is state-of-the-art in discovering high-fitness proteins with up to 8 mutations from the training set. We study the GFP and AAV design problems, ablations, and baselines to elucidate the results. Code: https://github.com/kirjner/GGS
Conformal Language Modeling
Jun 16, 2023We propose a novel approach to conformal prediction for generative language models (LMs). Standard conformal prediction produces prediction sets -- in place of single predictions -- that have rigorous, statistical performance guarantees. LM responses are typically sampled from the model's predicted distribution over the large, combinatorial output space of natural language. Translating this process to conformal prediction, we calibrate a stopping rule for sampling different outputs from the LM that get added to a growing set of candidates until we are confident that the output set is sufficient. Since some samples may be low-quality, we also simultaneously calibrate and apply a rejection rule for removing candidates from the output set to reduce noise. Similar to conformal prediction, we prove that the sampled set returned by our procedure contains at least one acceptable answer with high probability, while still being empirically precise (i.e., small) on average. Furthermore, within this set of candidate responses, we show that we can also accurately identify subsets of individual components -- such as phrases or sentences -- that are each independently correct (e.g., that are not "hallucinations"), again with statistical guarantees. We demonstrate the promise of our approach on multiple tasks in open-domain question answering, text summarization, and radiology report generation using different LM variants.
RxnScribe: A Sequence Generation Model for Reaction Diagram Parsing
May 19, 2023Reaction diagram parsing is the task of extracting reaction schemes from a diagram in the chemistry literature. The reaction diagrams can be arbitrarily complex, thus robustly parsing them into structured data is an open challenge. In this paper, we present RxnScribe, a machine learning model for parsing reaction diagrams of varying styles. We formulate this structured prediction task with a sequence generation approach, which condenses the traditional pipeline into an end-to-end model. We train RxnScribe on a dataset of 1,378 diagrams and evaluate it with cross validation, achieving an 80.0% soft match F1 score, with significant improvements over previous models. Our code and data are publicly available at https://github.com/thomas0809/RxnScribe.
DiffDock-PP: Rigid Protein-Protein Docking with Diffusion Models
Apr 08, 2023



Understanding how proteins structurally interact is crucial to modern biology, with applications in drug discovery and protein design. Recent machine learning methods have formulated protein-small molecule docking as a generative problem with significant performance boosts over both traditional and deep learning baselines. In this work, we propose a similar approach for rigid protein-protein docking: DiffDock-PP is a diffusion generative model that learns to translate and rotate unbound protein structures into their bound conformations. We achieve state-of-the-art performance on DIPS with a median C-RMSD of 4.85, outperforming all considered baselines. Additionally, DiffDock-PP is faster than all search-based methods and generates reliable confidence estimates for its predictions. Our code is publicly available at $\texttt{https://github.com/ketatam/DiffDock-PP}$
PEOPL: Characterizing Privately Encoded Open Datasets with Public Labels
Mar 31, 2023



Allowing organizations to share their data for training of machine learning (ML) models without unintended information leakage is an open problem in practice. A promising technique for this still-open problem is to train models on the encoded data. Our approach, called Privately Encoded Open Datasets with Public Labels (PEOPL), uses a certain class of randomly constructed transforms to encode sensitive data. Organizations publish their randomly encoded data and associated raw labels for ML training, where training is done without knowledge of the encoding realization. We investigate several important aspects of this problem: We introduce information-theoretic scores for privacy and utility, which quantify the average performance of an unfaithful user (e.g., adversary) and a faithful user (e.g., model developer) that have access to the published encoded data. We then theoretically characterize primitives in building families of encoding schemes that motivate the use of random deep neural networks. Empirically, we compare the performance of our randomized encoding scheme and a linear scheme to a suite of computational attacks, and we also show that our scheme achieves competitive prediction accuracy to raw-sample baselines. Moreover, we demonstrate that multiple institutions, using independent random encoders, can collaborate to train improved ML models.