 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeExpectile Neural Networks for Genetic Data Analysis of Complex Diseases
Oct 26, 2020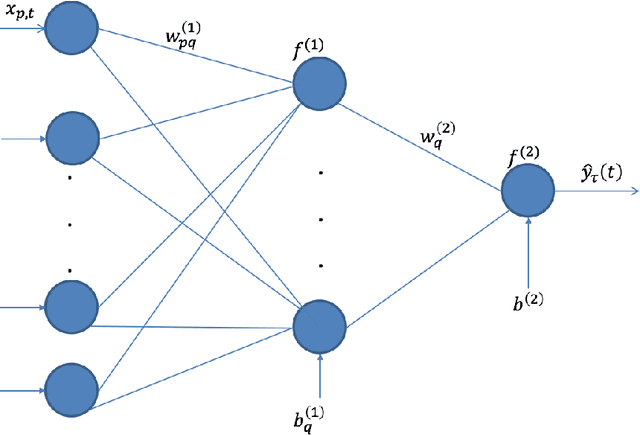
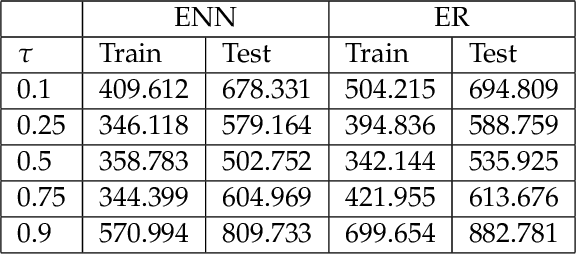
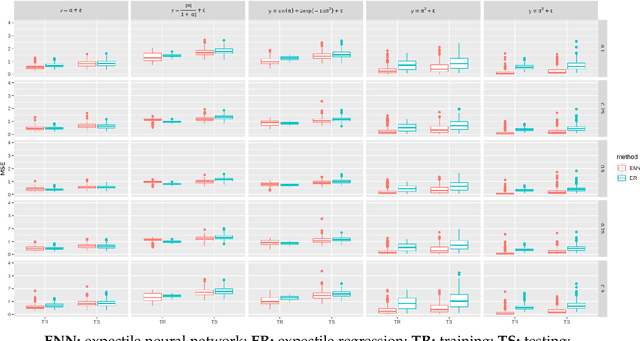
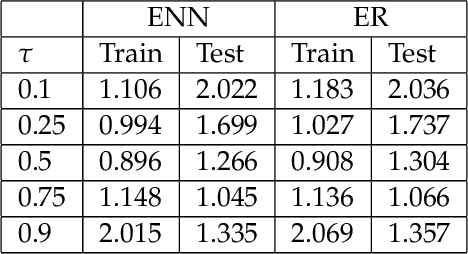
The genetic etiologies of common diseases are highly complex and heterogeneous. Classic statistical methods, such as linear regression, have successfully identified numerous genetic variants associated with complex diseases. Nonetheless, for most complex diseases, the identified variants only account for a small proportion of heritability. Challenges remain to discover additional variants contributing to complex diseases. Expectile regression is a generalization of linear regression and provides completed information on the conditional distribution of a phenotype of interest. While expectile regression has many nice properties and holds great promise for genetic data analyses (e.g., investigating genetic variants predisposing to a high-risk population), it has been rarely used in genetic research. In this paper, we develop an expectile neural network (ENN) method for genetic data analyses of complex diseases. Similar to expectile regression, ENN provides a comprehensive view of relationships between genetic variants and disease phenotypes and can be used to discover genetic variants predisposing to sub-populations (e.g., high-risk groups). We further integrate the idea of neural networks into ENN, making it capable of capturing non-linear and non-additive genetic effects (e.g., gene-gene interactions). Through simulations, we showed that the proposed method outperformed an existing expectile regression when there exist complex relationships between genetic variants and disease phenotypes. We also applied the proposed method to the genetic data from the Study of Addiction: Genetics and Environment(SAGE), investigating the relationships of candidate genes with smoking quantity.
NASS: Optimizing Secure Inference via Neural Architecture Search
Feb 16, 2020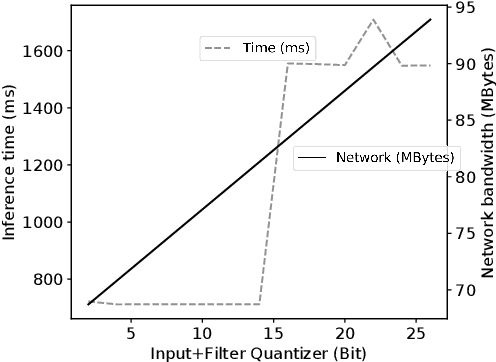
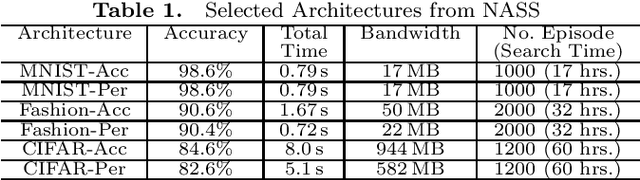
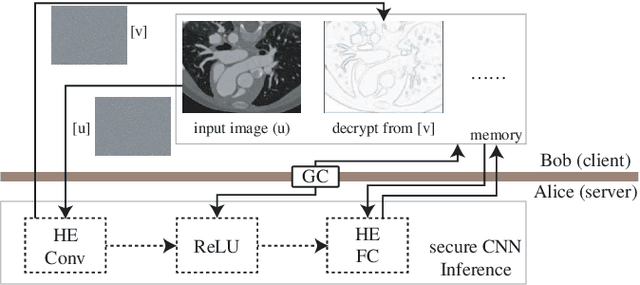
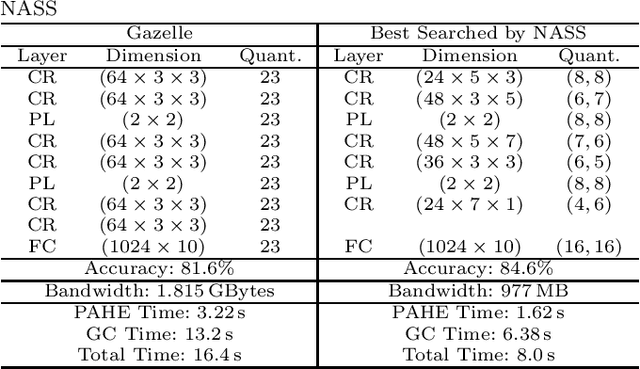
Due to increasing privacy concerns, neural network (NN) based secure inference (SI) schemes that simultaneously hide the client inputs and server models attract major research interests. While existing works focused on developing secure protocols for NN-based SI, in this work, we take a different approach. We propose NASS, an integrated framework to search for tailored NN architectures designed specifically for SI. In particular, we propose to model cryptographic protocols as design elements with associated reward functions. The characterized models are then adopted in a joint optimization with predicted hyperparameters in identifying the best NN architectures that balance prediction accuracy and execution efficiency. In the experiment, it is demonstrated that we can achieve the best of both worlds by using NASS, where the prediction accuracy can be improved from 81.6% to 84.6%, while the inference runtime is reduced by 2x and communication bandwidth by 1.9x on the CIFAR-10 dataset.
On Neural Architecture Search for Resource-Constrained Hardware Platforms
Oct 31, 2019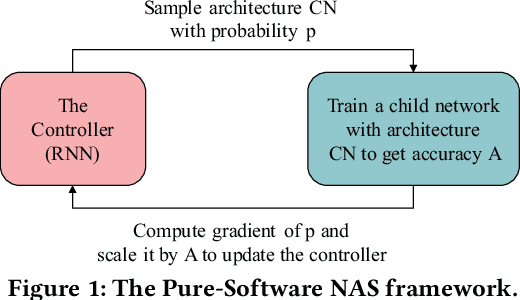
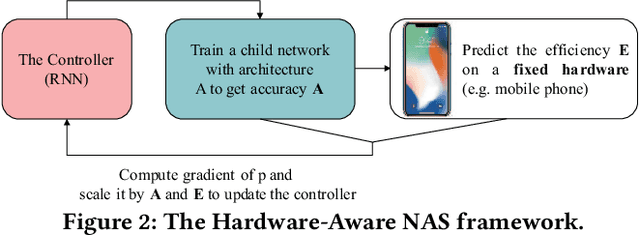
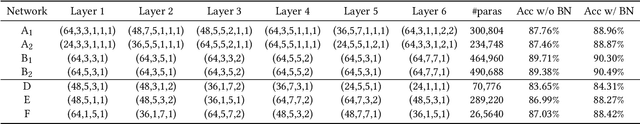
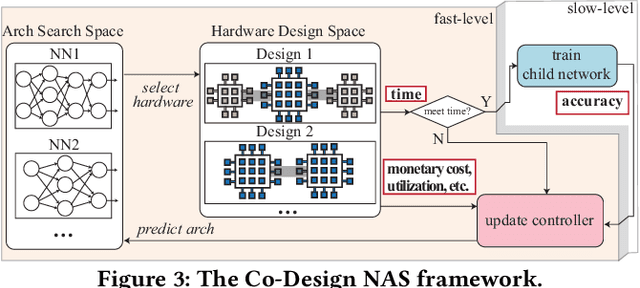
In the recent past, the success of Neural Architecture Search (NAS) has enabled researchers to broadly explore the design space using learning-based methods. Apart from finding better neural network architectures, the idea of automation has also inspired to improve their implementations on hardware. While some practices of hardware machine-learning automation have achieved remarkable performance, the traditional design concept is still followed: a network architecture is first structured with excellent test accuracy, and then compressed and optimized to fit into a target platform. Such a design flow will easily lead to inferior local-optimal solutions. To address this problem, we propose a new framework to jointly explore the space of neural architecture, hardware implementation, and quantization. Our objective is to find a quantized architecture with the highest accuracy that is implementable on given hardware specifications. We employ FPGAs to implement and test our designs with limited loop-up tables (LUTs) and required throughput. Compared to the separate design/searching methods, our framework has demonstrated much better performance under strict specifications and generated designs of higher accuracy by 18\% to 68\% in the task of classifying CIFAR10 images. With 30,000 LUTs, a light-weight design is found to achieve 82.98\% accuracy and 1293 images/second throughput, compared to which, under the same constraints, the traditional method even fails to find a valid solution.
Grand Challenge of 106-Point Facial Landmark Localization
May 09, 2019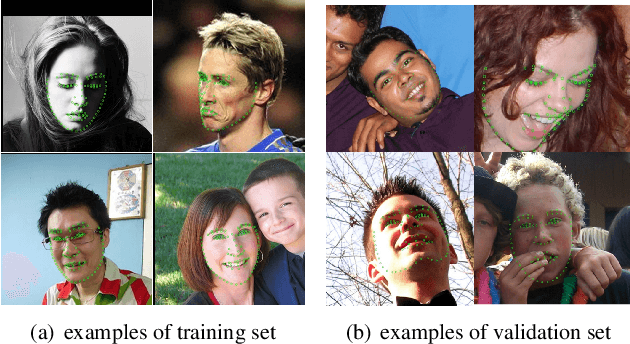
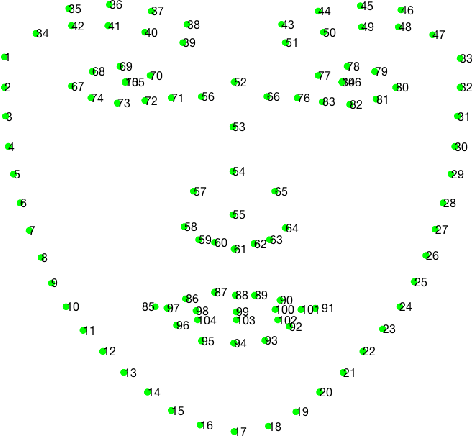

Facial landmark localization is a very crucial step in numerous face related applications, such as face recognition, facial pose estimation, face image synthesis, etc. However, previous competitions on facial landmark localization (i.e., the 300-W, 300-VW and Menpo challenges) aim to predict 68-point landmarks, which are incompetent to depict the structure of facial components. In order to overcome this problem, we construct a challenging dataset, named JD-landmark. Each image is manually annotated with 106-point landmarks. This dataset covers large variations on pose and expression, which brings a lot of difficulties to predict accurate landmarks. We hold a 106-point facial landmark localization competition1 on this dataset in conjunction with IEEE International Conference on Multimedia and Expo (ICME) 2019. The purpose of this competition is to discover effective and robust facial landmark localization approaches.
Quantization of Fully Convolutional Networks for Accurate Biomedical Image Segmentation
Mar 13, 2018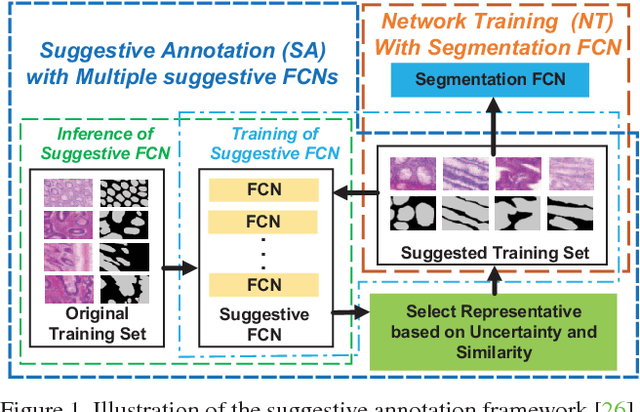
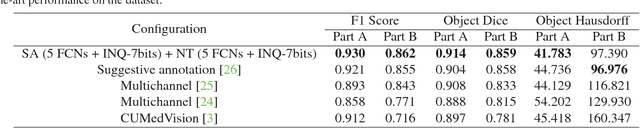
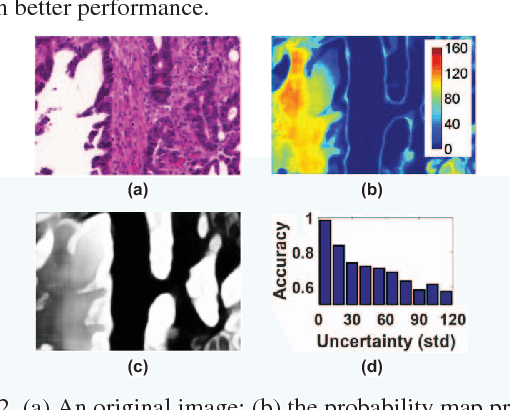
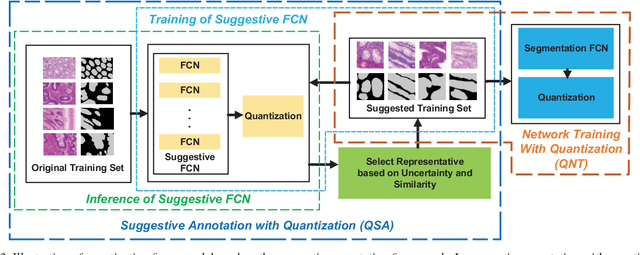
With pervasive applications of medical imaging in health-care, biomedical image segmentation plays a central role in quantitative analysis, clinical diagno- sis, and medical intervention. Since manual anno- tation su ers limited reproducibility, arduous e orts, and excessive time, automatic segmentation is desired to process increasingly larger scale histopathological data. Recently, deep neural networks (DNNs), par- ticularly fully convolutional networks (FCNs), have been widely applied to biomedical image segmenta- tion, attaining much improved performance. At the same time, quantization of DNNs has become an ac- tive research topic, which aims to represent weights with less memory (precision) to considerably reduce memory and computation requirements of DNNs while maintaining acceptable accuracy. In this paper, we apply quantization techniques to FCNs for accurate biomedical image segmentation. Unlike existing litera- ture on quantization which primarily targets memory and computation complexity reduction, we apply quan- tization as a method to reduce over tting in FCNs for better accuracy. Speci cally, we focus on a state-of- the-art segmentation framework, suggestive annotation [22], which judiciously extracts representative annota- tion samples from the original training dataset, obtain- ing an e ective small-sized balanced training dataset. We develop two new quantization processes for this framework: (1) suggestive annotation with quantiza- tion for highly representative training samples, and (2) network training with quantization for high accuracy. Extensive experiments on the MICCAI Gland dataset show that both quantization processes can improve the segmentation performance, and our proposed method exceeds the current state-of-the-art performance by up to 1%. In addition, our method has a reduction of up to 6.4x on memory usage.
Generalized Similarity U: A Non-parametric Test of Association Based on Similarity
Jan 04, 2018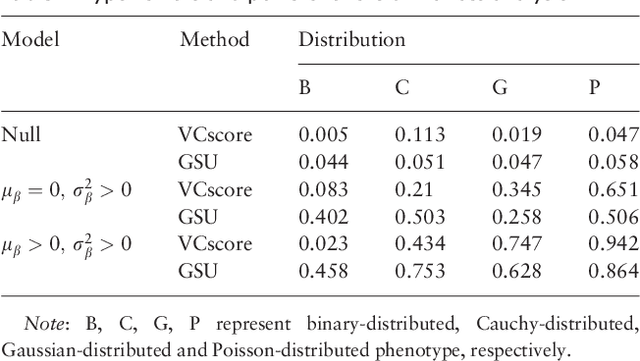
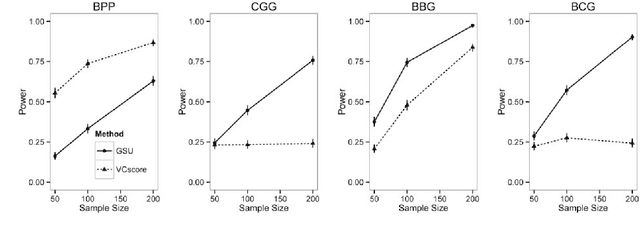
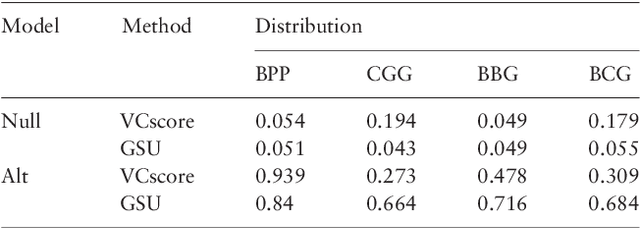
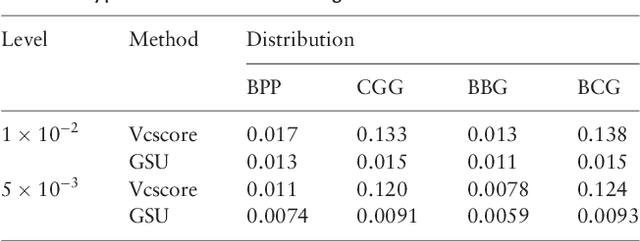
Second generation sequencing technologies are being increasingly used for genetic association studies, where the main research interest is to identify sets of genetic variants that contribute to various phenotype. The phenotype can be univariate disease status, multivariate responses and even high-dimensional outcomes. Considering the genotype and phenotype as two complex objects, this also poses a general statistical problem of testing association between complex objects. We here proposed a similarity-based test, generalized similarity U (GSU), that can test the association between complex objects. We first studied the theoretical properties of the test in a general setting and then focused on the application of the test to sequencing association studies. Based on theoretical analysis, we proposed to use Laplacian kernel based similarity for GSU to boost power and enhance robustness. Through simulation, we found that GSU did have advantages over existing methods in terms of power and robustness. We further performed a whole genome sequencing (WGS) scan for Alzherimer Disease Neuroimaging Initiative (ADNI) data, identifying three genes, APOE, APOC1 and TOMM40, associated with imaging phenotype. We developed a C++ package for analysis of whole genome sequencing data using GSU. The source codes can be downloaded at https://github.com/changshuaiwei/gsu.
Trees Assembling Mann Whitney Approach for Detecting Genome-wide Joint Association among Low Marginal Effect loci
May 05, 2015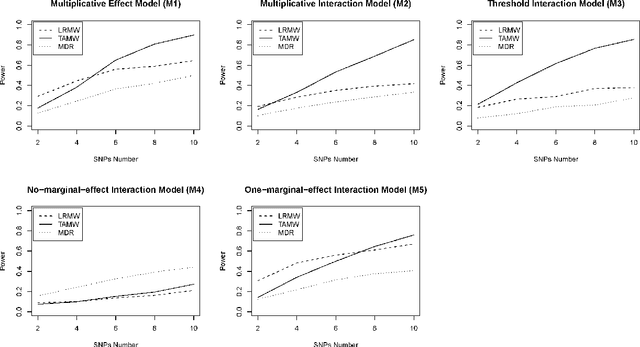
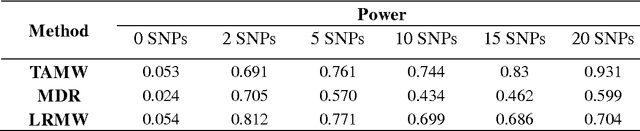
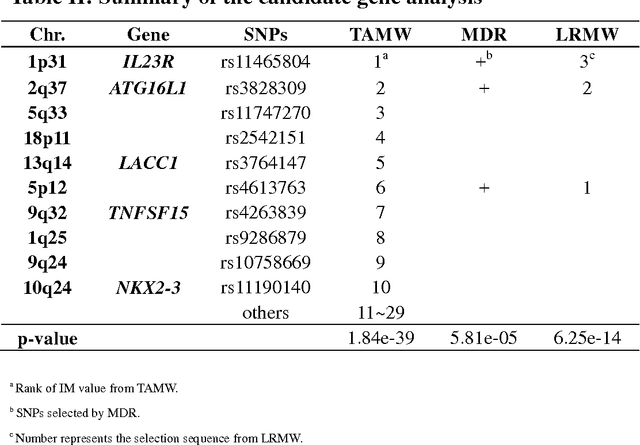
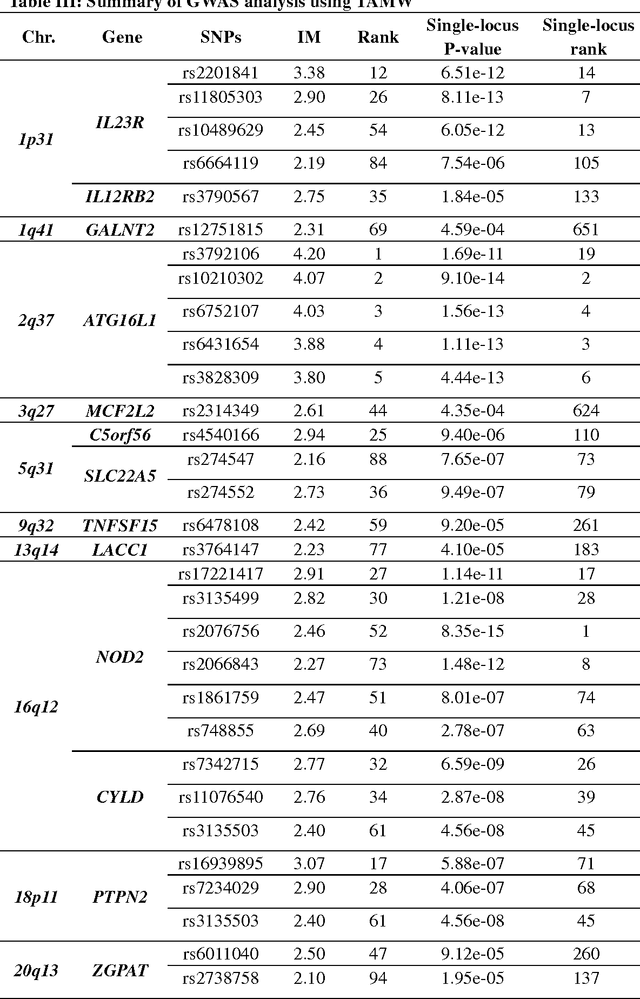
Common complex diseases are likely influenced by the interplay of hundreds, or even thousands, of genetic variants. Converging evidence shows that genetic variants with low marginal effects (LME) play an important role in disease development. Despite their potential significance, discovering LME genetic variants and assessing their joint association on high dimensional data (e.g., genome wide association studies) remain a great challenge. To facilitate joint association analysis among a large ensemble of LME genetic variants, we proposed a computationally efficient and powerful approach, which we call Trees Assembling Mann whitney (TAMW). Through simulation studies and an empirical data application, we found that TAMW outperformed multifactor dimensionality reduction (MDR) and the likelihood ratio based Mann whitney approach (LRMW) when the underlying complex disease involves multiple LME loci and their interactions. For instance, in a simulation with 20 interacting LME loci, TAMW attained a higher power (power=0.931) than both MDR (power=0.599) and LRMW (power=0.704). In an empirical study of 29 known Crohn's disease (CD) loci, TAMW also identified a stronger joint association with CD than those detected by MDR and LRMW. Finally, we applied TAMW to Wellcome Trust CD GWAS to conduct a genome wide analysis. The analysis of 459K single nucleotide polymorphisms was completed in 40 hours using parallel computing, and revealed a joint association predisposing to CD (p-value=2.763e-19). Further analysis of the newly discovered association suggested that 13 genes, such as ATG16L1 and LACC1, may play an important role in CD pathophysiological and etiological processes.