 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeALBRT: Cellular Composition Prediction in Routine Histology Images
Aug 26, 2021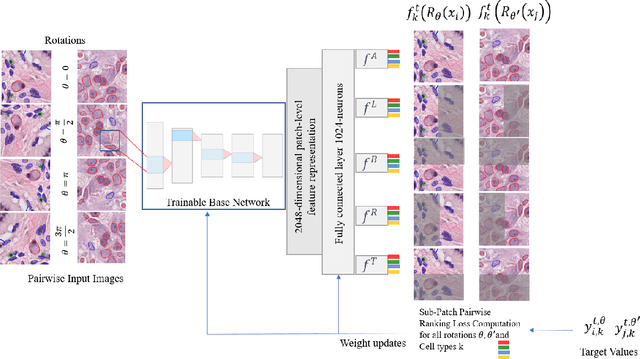
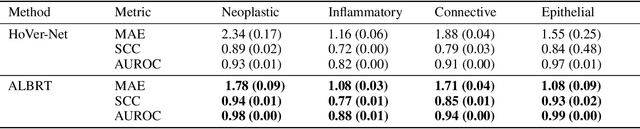
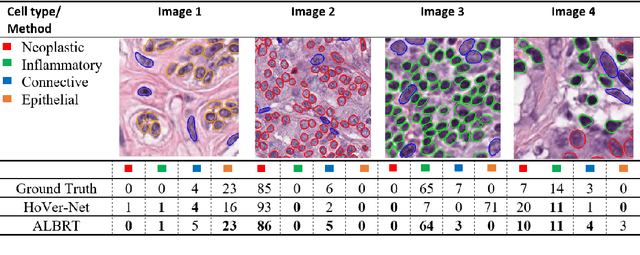
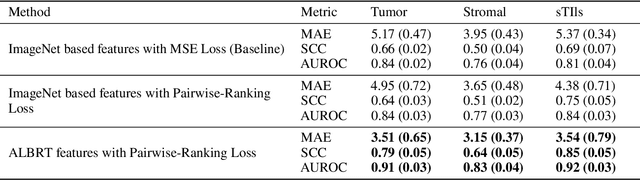
Cellular composition prediction, i.e., predicting the presence and counts of different types of cells in the tumor microenvironment from a digitized image of a Hematoxylin and Eosin (H&E) stained tissue section can be used for various tasks in computational pathology such as the analysis of cellular topology and interactions, subtype prediction, survival analysis, etc. In this work, we propose an image-based cellular composition predictor (ALBRT) which can accurately predict the presence and counts of different types of cells in a given image patch. ALBRT, by its contrastive-learning inspired design, learns a compact and rotation-invariant feature representation that is then used for cellular composition prediction of different cell types. It offers significant improvement over existing state-of-the-art approaches for cell classification and counting. The patch-level feature representation learned by ALBRT is transferrable for cellular composition analysis over novel datasets and can also be utilized for downstream prediction tasks in CPath as well. The code and the inference webserver for the proposed method are available at the URL: https://github.com/engrodawood/ALBRT.
All You Need is Color: Image based Spatial Gene Expression Prediction using Neural Stain Learning
Aug 26, 2021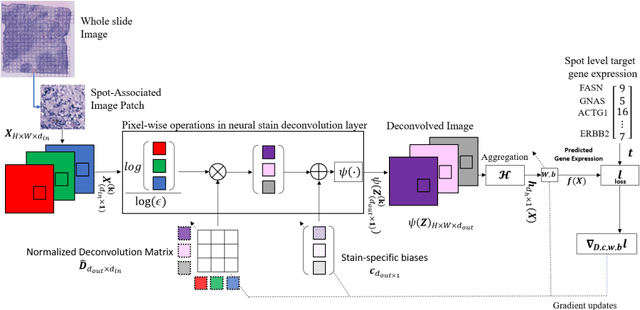
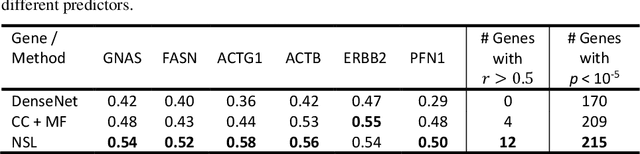
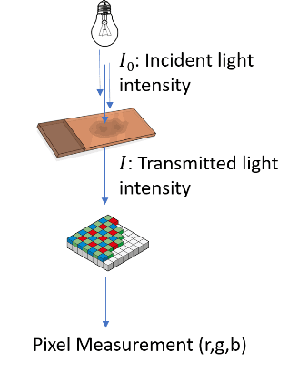
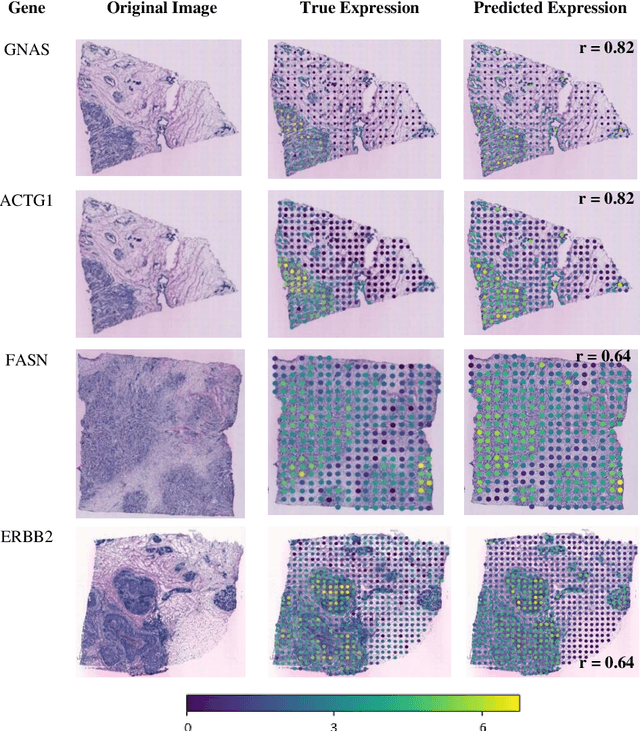
"Is it possible to predict expression levels of different genes at a given spatial location in the routine histology image of a tumor section by modeling its stain absorption characteristics?" In this work, we propose a "stain-aware" machine learning approach for prediction of spatial transcriptomic gene expression profiles using digital pathology image of a routine Hematoxylin & Eosin (H&E) histology section. Unlike recent deep learning methods which are used for gene expression prediction, our proposed approach termed Neural Stain Learning (NSL) explicitly models the association of stain absorption characteristics of the tissue with gene expression patterns in spatial transcriptomics by learning a problem-specific stain deconvolution matrix in an end-to-end manner. The proposed method with only 11 trainable weight parameters outperforms both classical regression models with cellular composition and morphological features as well as deep learning methods. We have found that the gene expression predictions from the proposed approach show higher correlations with true expression values obtained through sequencing for a larger set of genes in comparison to other approaches.
L1-regularized neural ranking for risk stratification and its application to prediction of time to distant metastasis in luminal node negative chemotherapy naïve breast cancer patients
Aug 23, 2021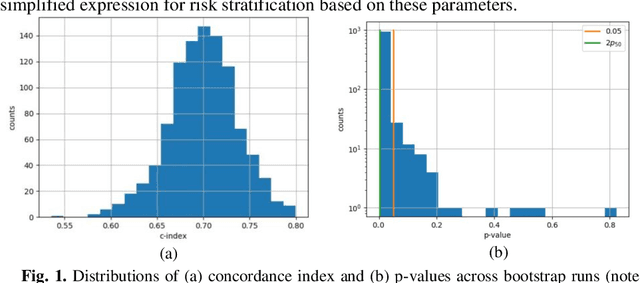

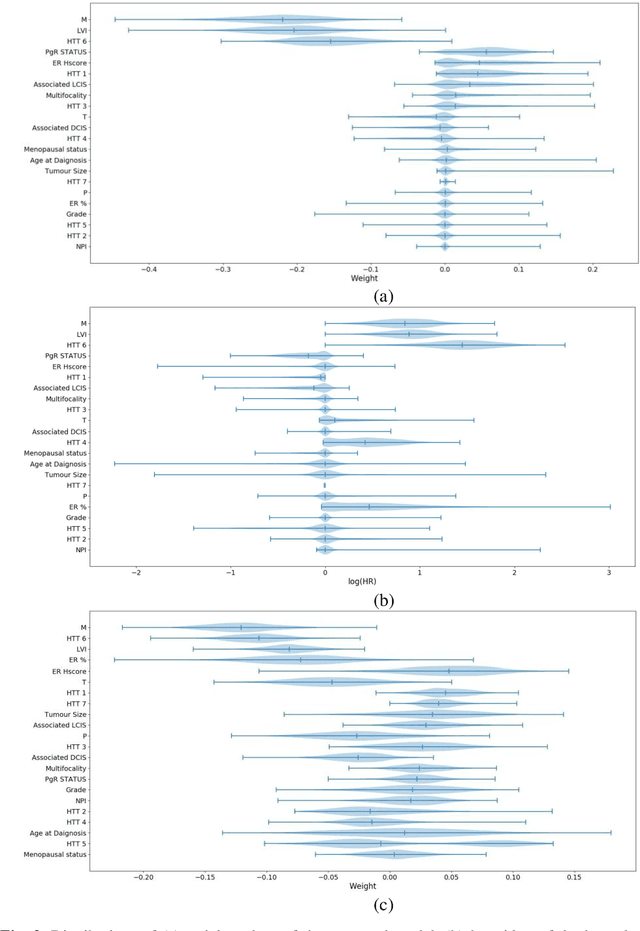
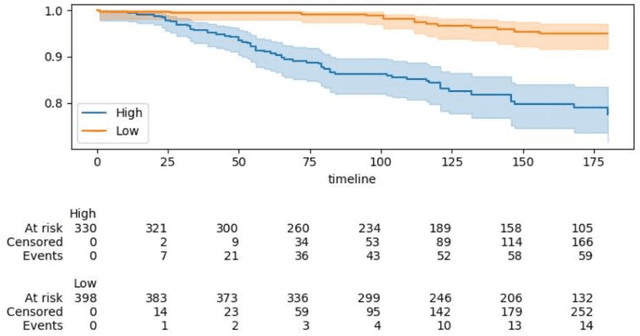
Can we predict if an early stage cancer patient is at high risk of developing distant metastasis and what clinicopathological factors are associated with such a risk? In this paper, we propose a ranking based censoring-aware machine learning model for answering such questions. The proposed model is able to generate an interpretable formula for risk stratifi-cation using a minimal number of clinicopathological covariates through L1-regulrization. Using this approach, we analyze the association of time to distant metastasis (TTDM) with various clinical parameters for early stage, luminal (ER+ or HER2-) breast cancer patients who received endocrine therapy but no chemotherapy (n = 728). The TTDM risk stratification formula obtained using the proposed approach is primarily based on mitotic score, histolog-ical tumor type and lymphovascular invasion. These findings corroborate with the known role of these covariates in increased risk for distant metastasis. Our analysis shows that the proposed risk stratification formula can discriminate between cases with high and low risk of distant metastasis (p-value < 0.005) and can also rank cases based on their time to distant metastasis with a concordance-index of 0.73.
HydraMix-Net: A Deep Multi-task Semi-supervised Learning Approach for Cell Detection and Classification
Aug 11, 2020Semi-supervised techniques have removed the barriers of large scale labelled set by exploiting unlabelled data to improve the performance of a model. In this paper, we propose a semi-supervised deep multi-task classification and localization approach HydraMix-Net in the field of medical imagining where labelling is time consuming and costly. Firstly, the pseudo labels are generated using the model's prediction on the augmented set of unlabelled image with averaging. The high entropy predictions are further sharpened to reduced the entropy and are then mixed with the labelled set for training. The model is trained in multi-task learning manner with noise tolerant joint loss for classification localization and achieves better performance when given limited data in contrast to a simple deep model. On DLBCL data it achieves 80\% accuracy in contrast to simple CNN achieving 70\% accuracy when given only 100 labelled examples.
Context-Aware Convolutional Neural Network for Grading of Colorectal Cancer Histology Images
Jul 22, 2019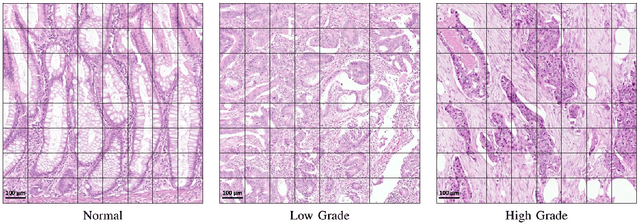
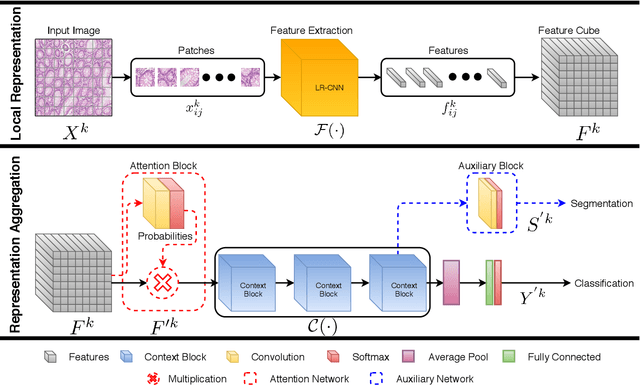

Digital histology images are amenable to the application of convolutional neural network (CNN) for analysis due to the sheer size of pixel data present in them. CNNs are generally used for representation learning from small image patches (e.g. 224x224) extracted from digital histology images due to computational and memory constraints. However, this approach does not incorporate high-resolution contextual information in histology images. We propose a novel way to incorporate larger context by a context-aware neural network based on images with a dimension of 1,792x1,792 pixels. The proposed framework first encodes the local representation of a histology image into high dimensional features then aggregates the features by considering their spatial organization to make a final prediction. The proposed method is evaluated for colorectal cancer grading and breast cancer classification. A comprehensive analysis of some variants of the proposed method is presented. Our method outperformed the traditional patch-based approaches, problem-specific methods, and existing context-based methods quantitatively by a margin of 3.61%. Code and dataset related information is available at this link: https://tia-lab.github.io/Context-Aware-CNN
Learning Where to See: A Novel Attention Model for Automated Immunohistochemical Scoring
Mar 26, 2019



Estimating over-amplification of human epidermal growth factor receptor 2 (HER2) on invasive breast cancer (BC) is regarded as a significant predictive and prognostic marker. We propose a novel deep reinforcement learning (DRL) based model that treats immunohistochemical (IHC) scoring of HER2 as a sequential learning task. For a given image tile sampled from multi-resolution giga-pixel whole slide image (WSI), the model learns to sequentially identify some of the diagnostically relevant regions of interest (ROIs) by following a parameterized policy. The selected ROIs are processed by recurrent and residual convolution networks to learn the discriminative features for different HER2 scores and predict the next location, without requiring to process all the sub-image patches of a given tile for predicting the HER2 score, mimicking the histopathologist who would not usually analyze every part of the slide at the highest magnification. The proposed model incorporates a task-specific regularization term and inhibition of return mechanism to prevent the model from revisiting the previously attended locations. We evaluated our model on two IHC datasets: a publicly available dataset from the HER2 scoring challenge contest and another dataset consisting of WSIs of gastroenteropancreatic neuroendocrine tumor sections stained with Glo1 marker. We demonstrate that the proposed model outperforms other methods based on state-of-the-art deep convolutional networks. To the best of our knowledge, this is the first study using DRL for IHC scoring and could potentially lead to wider use of DRL in the domain of computational pathology reducing the computational burden of the analysis of large multigigapixel histology images.
Micro-Net: A unified model for segmentation of various objects in microscopy images
Apr 22, 2018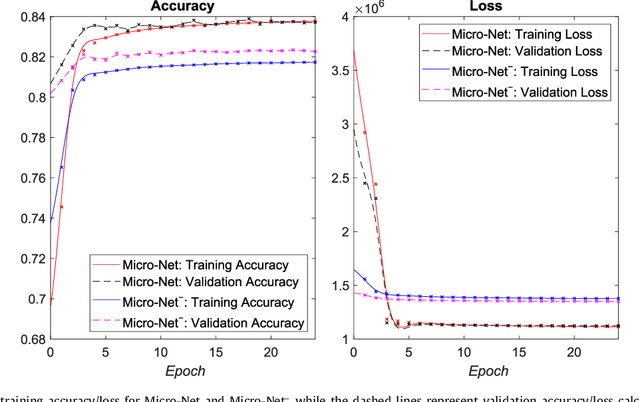
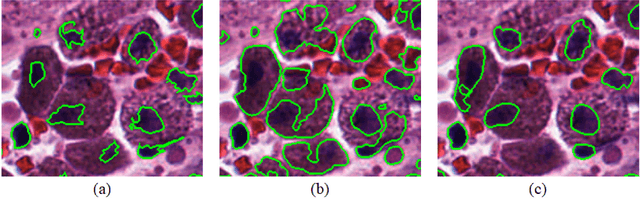
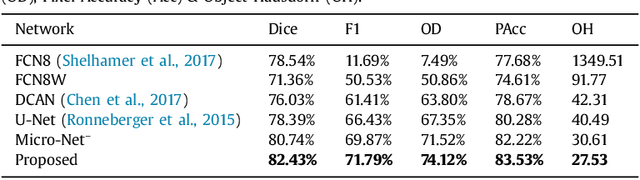
Object segmentation and structure localization are important steps in automated image analysis pipelines for microscopy images. We present a convolution neural network (CNN) based deep learning architecture for segmentation of objects in microscopy images. The proposed network can be used to segment cells, nuclei and glands in fluorescence microscopy and histology images after slight tuning of its parameters. It trains itself at multiple resolutions of the input image, connects the intermediate layers for better localization and context and generates the output using multi-resolution deconvolution filters. The extra convolutional layers which bypass the max-pooling operation allow the network to train for variable input intensities and object size and make it robust to noisy data. We compare our results on publicly available data sets and show that the proposed network outperforms the state-of-the-art.
Representation-Aggregation Networks for Segmentation of Multi-Gigapixel Histology Images
Jul 27, 2017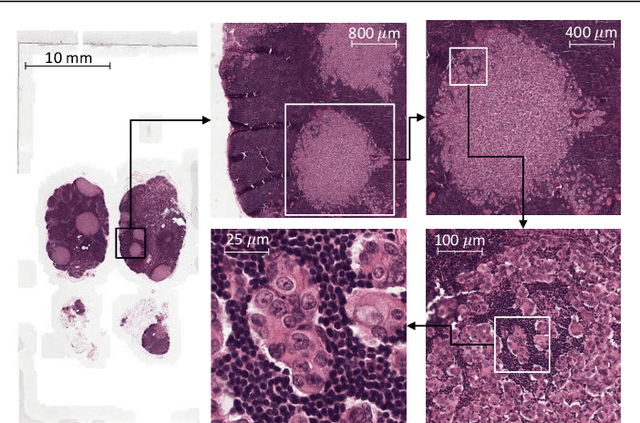
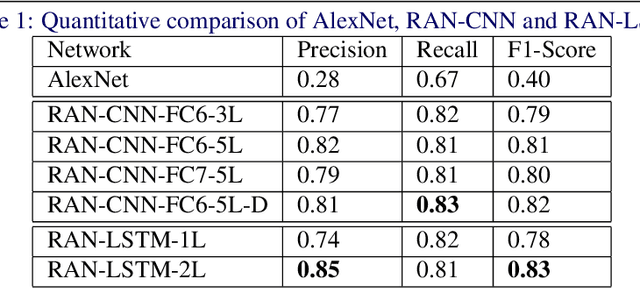
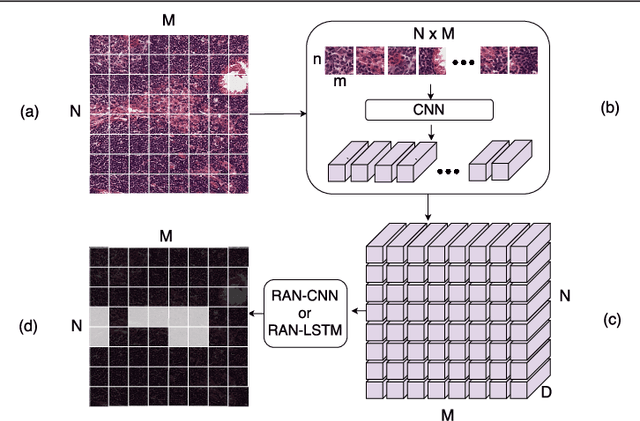
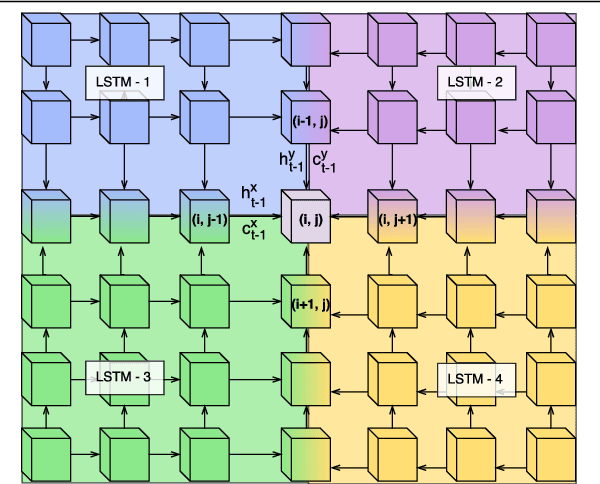
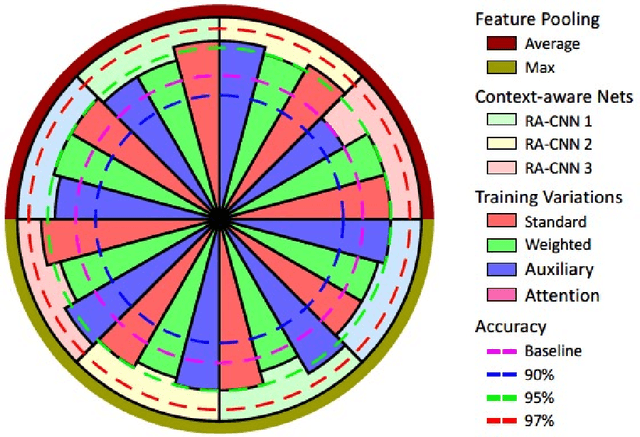
Convolutional Neural Network (CNN) models have become the state-of-the-art for most computer vision tasks with natural images. However, these are not best suited for multi-gigapixel resolution Whole Slide Images (WSIs) of histology slides due to large size of these images. Current approaches construct smaller patches from WSIs which results in the loss of contextual information. We propose to capture the spatial context using novel Representation-Aggregation Network (RAN) for segmentation purposes, wherein the first network learns patch-level representation and the second network aggregates context from a grid of neighbouring patches. We can use any CNN for representation learning, and can utilize CNN or 2D-Long Short Term Memory (2D-LSTM) for context-aggregation. Our method significantly outperformed conventional patch-based CNN approaches on segmentation of tumour in WSIs of breast cancer tissue sections.
Gland Segmentation in Colon Histology Images: The GlaS Challenge Contest
Sep 01, 2016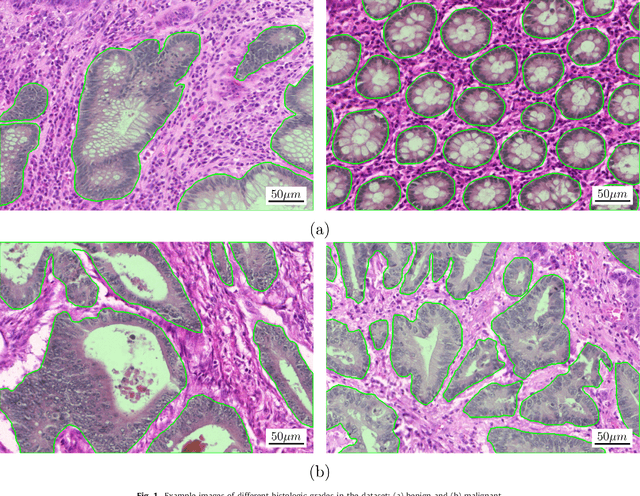
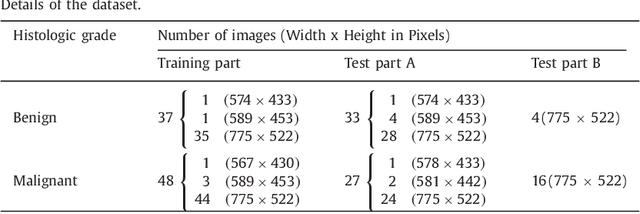
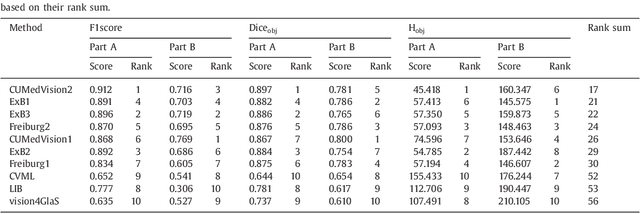
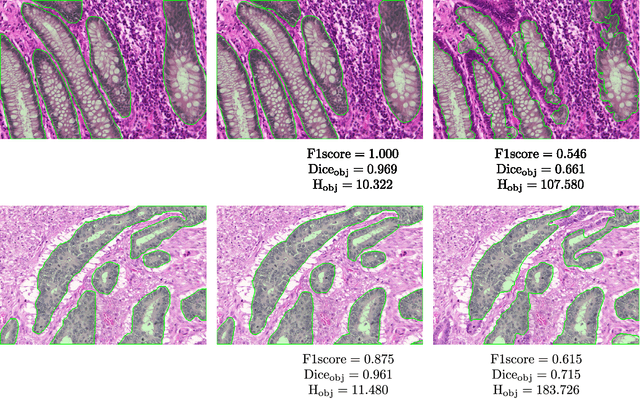
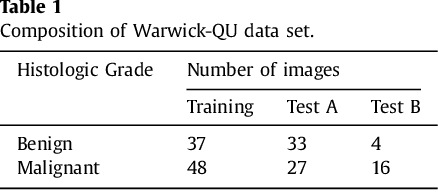
Colorectal adenocarcinoma originating in intestinal glandular structures is the most common form of colon cancer. In clinical practice, the morphology of intestinal glands, including architectural appearance and glandular formation, is used by pathologists to inform prognosis and plan the treatment of individual patients. However, achieving good inter-observer as well as intra-observer reproducibility of cancer grading is still a major challenge in modern pathology. An automated approach which quantifies the morphology of glands is a solution to the problem. This paper provides an overview to the Gland Segmentation in Colon Histology Images Challenge Contest (GlaS) held at MICCAI'2015. Details of the challenge, including organization, dataset and evaluation criteria, are presented, along with the method descriptions and evaluation results from the top performing methods.
Assessment of algorithms for mitosis detection in breast cancer histopathology images
Nov 21, 2014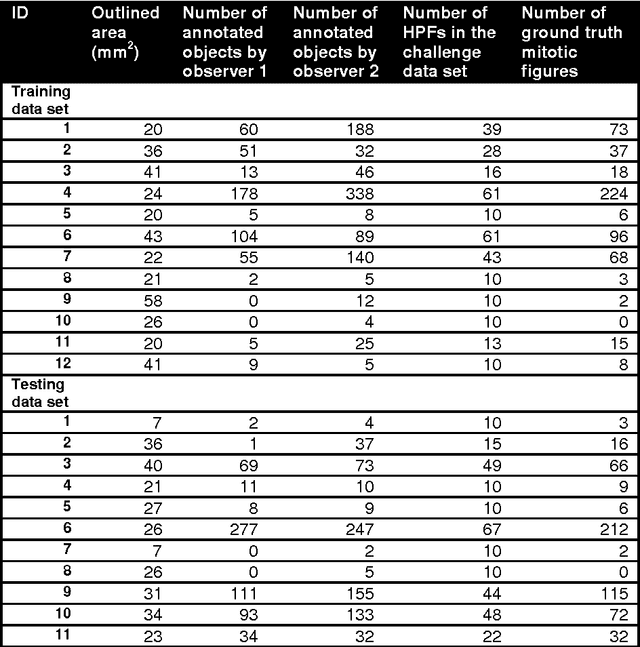

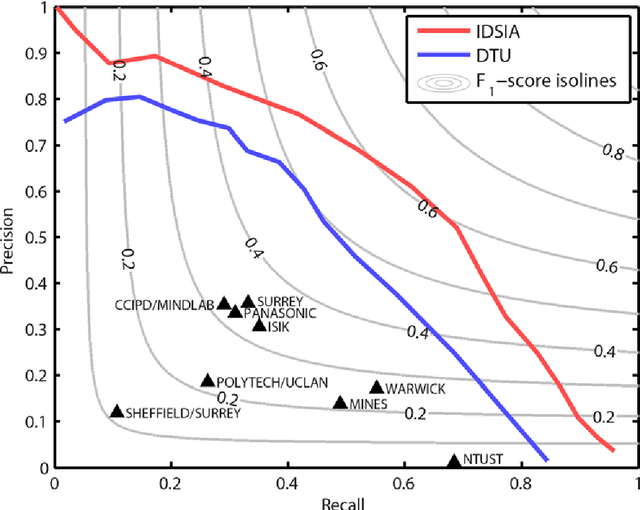
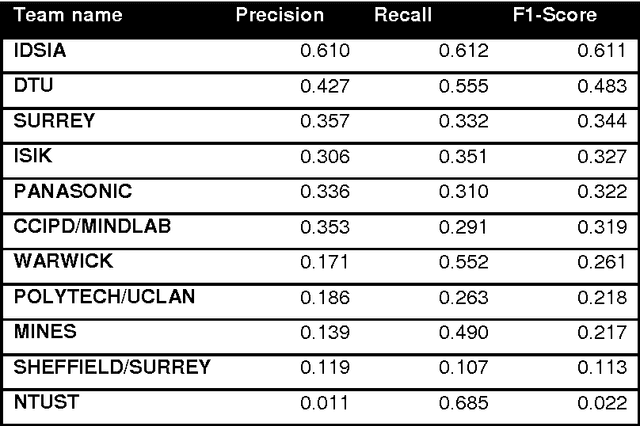
The proliferative activity of breast tumors, which is routinely estimated by counting of mitotic figures in hematoxylin and eosin stained histology sections, is considered to be one of the most important prognostic markers. However, mitosis counting is laborious, subjective and may suffer from low inter-observer agreement. With the wider acceptance of whole slide images in pathology labs, automatic image analysis has been proposed as a potential solution for these issues. In this paper, the results from the Assessment of Mitosis Detection Algorithms 2013 (AMIDA13) challenge are described. The challenge was based on a data set consisting of 12 training and 11 testing subjects, with more than one thousand annotated mitotic figures by multiple observers. Short descriptions and results from the evaluation of eleven methods are presented. The top performing method has an error rate that is comparable to the inter-observer agreement among pathologists.