 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeScaling Novel Graph Generation via Lightweight Structure-Guided Autoregressive Models
Jun 02, 2026Generating realistic and diverse graphs is a key problem in machine learning, with applications in molecular discovery, circuit design, cybersecurity, and beyond. However, current graph generative models remain limited by scalability and novelty. Diffusion-based methods often require costly full-adjacency operations and long denoising chains, while many autoregressive and hybrid models have at least quadratic complexity. In addition, these models often imitate training graphs rather than generalize beyond them. We propose a lightweight autoregressive framework to address these issues. It uses a structure-guided topological ordering to serialize graphs into regular edge sequences, enabling near log-linear generation, and a two-phase training strategy that combines exploration-oriented augmentation with iterative refinement to reduce overfitting and promote controlled novelty. Experiments on molecular and non-molecular benchmarks show that our approach improves novelty while preserving high validity and uniqueness. The framework also supports both LSTM and Mamba-style causal sequence backbones, with large-memory accelerators enabling longer graph-sequence experiments beyond typical GPU limits.
Scalable Heterogeneous Graph Foundation Models for Data-Driven Optimal Power Flow in Smart Grids
May 22, 2026Fast and reliable optimal power flow (OPF) approximation is essential for reliable smart-grid operation, yet many learning-based surrogates either flatten the native heterogeneous structure of power networks, target a limited set of grid topologies, or lack scalable infrastructure for graph foundation model (GFM) training. This paper presents a scalable heterogeneous graph neural network (GNN) workflow, built on HydraGNN, for data-driven OPF surrogate modeling and OPF-GFM development. The workflow preserves the distinct node and edge types of power grids -- buses, generators, loads, shunts, AC lines, transformers, and device-to-bus couplings -- and supports distributed preprocessing, training, hyperparameter optimization (HPO), and downstream fine-tuning on leadership-class supercomputers. Using three million heterogeneous graph instances spanning ten PGLib-OPF cases, from 14 to 13,659 buses, we conduct DeepHyper-driven HPO on the ORNL Frontier supercomputer. The campaign identifies compact models ($\sim$1.6--1.7M parameters) with the lowest validation losses. Downstream experiments on feasibility classification and N-1 contingency regression show that fine-tuning pretrained OPF GFM improves low-data accuracy, stabilizes training, accelerates convergence, and reduces adaptation cost when partial or head-only fine-tuning is used.
Scaling Laws of Graph Neural Networks for Atomistic Materials Modeling
Apr 10, 2025Atomistic materials modeling is a critical task with wide-ranging applications, from drug discovery to materials science, where accurate predictions of the target material property can lead to significant advancements in scientific discovery. Graph Neural Networks (GNNs) represent the state-of-the-art approach for modeling atomistic material data thanks to their capacity to capture complex relational structures. While machine learning performance has historically improved with larger models and datasets, GNNs for atomistic materials modeling remain relatively small compared to large language models (LLMs), which leverage billions of parameters and terabyte-scale datasets to achieve remarkable performance in their respective domains. To address this gap, we explore the scaling limits of GNNs for atomistic materials modeling by developing a foundational model with billions of parameters, trained on extensive datasets in terabyte-scale. Our approach incorporates techniques from LLM libraries to efficiently manage large-scale data and models, enabling both effective training and deployment of these large-scale GNN models. This work addresses three fundamental questions in scaling GNNs: the potential for scaling GNN model architectures, the effect of dataset size on model accuracy, and the applicability of LLM-inspired techniques to GNN architectures. Specifically, the outcomes of this study include (1) insights into the scaling laws for GNNs, highlighting the relationship between model size, dataset volume, and accuracy, (2) a foundational GNN model optimized for atomistic materials modeling, and (3) a GNN codebase enhanced with advanced LLM-based training techniques. Our findings lay the groundwork for large-scale GNNs with billions of parameters and terabyte-scale datasets, establishing a scalable pathway for future advancements in atomistic materials modeling.
Scalable Training of Graph Foundation Models for Atomistic Materials Modeling: A Case Study with HydraGNN
Jun 12, 2024We present our work on developing and training scalable graph foundation models (GFM) using HydraGNN, a multi-headed graph convolutional neural network architecture. HydraGNN expands the boundaries of graph neural network (GNN) in both training scale and data diversity. It abstracts over message passing algorithms, allowing both reproduction of and comparison across algorithmic innovations that define convolution in GNNs. This work discusses a series of optimizations that have allowed scaling up the GFM training to tens of thousands of GPUs on datasets that consist of hundreds of millions of graphs. Our GFMs use multi-task learning (MTL) to simultaneously learn graph-level and node-level properties of atomistic structures, such as the total energy and atomic forces. Using over 150 million atomistic structures for training, we illustrate the performance of our approach along with the lessons learned on two United States Department of Energy (US-DOE) supercomputers, namely the Perlmutter petascale system at the National Energy Research Scientific Computing Center and the Frontier exascale system at Oak Ridge National Laboratory. The HydraGNN architecture enables the GFM to achieve near-linear strong scaling performance using more than 2,000 GPUs on Perlmutter and 16,000 GPUs on Frontier. Hyperparameter optimization (HPO) was performed on over 64,000 GPUs on Frontier to select GFM architectures with high accuracy. Early stopping was applied on each GFM architecture for energy awareness in performing such an extreme-scale task. The training of an ensemble of highest-ranked GFM architectures continued until convergence to establish uncertainty quantification (UQ) capabilities with ensemble learning. Our contribution opens the door for rapidly developing, training, and deploying GFMs using large-scale computational resources to enable AI-accelerated materials discovery and design.
DeepSpeed4Science Initiative: Enabling Large-Scale Scientific Discovery through Sophisticated AI System Technologies
Oct 11, 2023



In the upcoming decade, deep learning may revolutionize the natural sciences, enhancing our capacity to model and predict natural occurrences. This could herald a new era of scientific exploration, bringing significant advancements across sectors from drug development to renewable energy. To answer this call, we present DeepSpeed4Science initiative (deepspeed4science.ai) which aims to build unique capabilities through AI system technology innovations to help domain experts to unlock today's biggest science mysteries. By leveraging DeepSpeed's current technology pillars (training, inference and compression) as base technology enablers, DeepSpeed4Science will create a new set of AI system technologies tailored for accelerating scientific discoveries by addressing their unique complexity beyond the common technical approaches used for accelerating generic large language models (LLMs). In this paper, we showcase the early progress we made with DeepSpeed4Science in addressing two of the critical system challenges in structural biology research.
A deep learning approach for detection and localization of leaf anomalies
Oct 07, 2022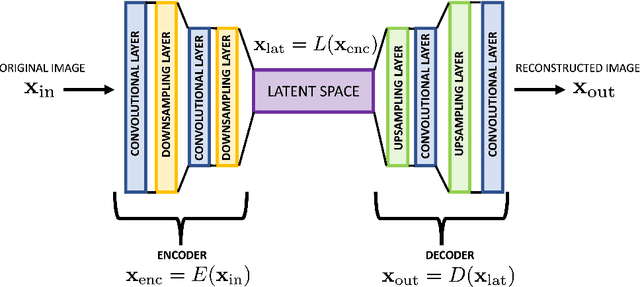
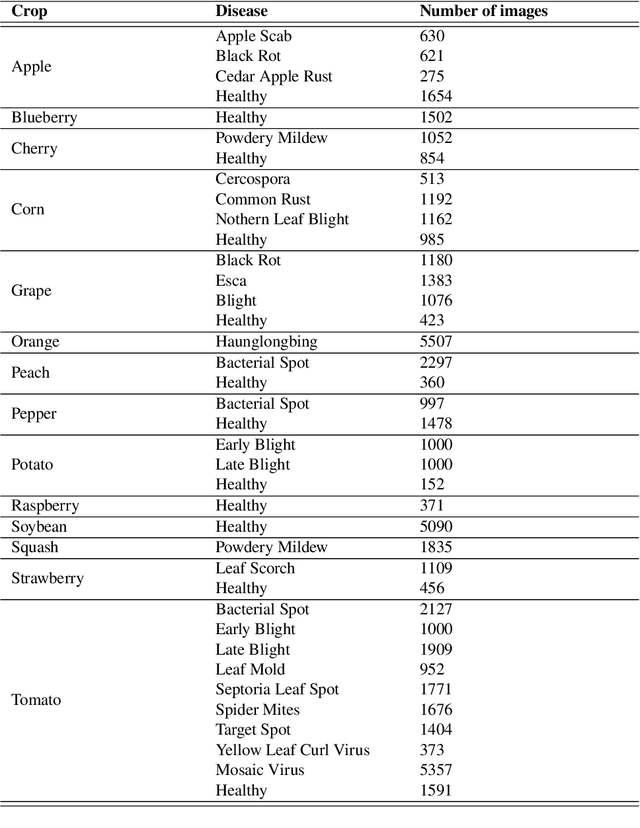
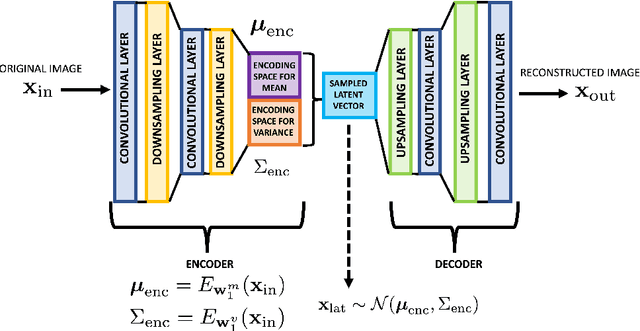
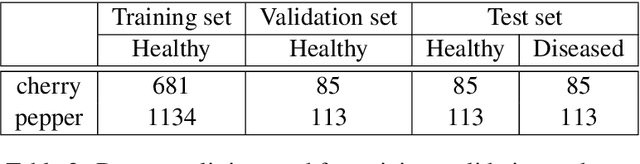
The detection and localization of possible diseases in crops are usually automated by resorting to supervised deep learning approaches. In this work, we tackle these goals with unsupervised models, by applying three different types of autoencoders to a specific open-source dataset of healthy and unhealthy pepper and cherry leaf images. CAE, CVAE and VQ-VAE autoencoders are deployed to screen unlabeled images of such a dataset, and compared in terms of image reconstruction, anomaly removal, detection and localization. The vector-quantized variational architecture turns out to be the best performing one with respect to all these targets.
A deep learning approach to solve forward differential problems on graphs
Oct 07, 2022
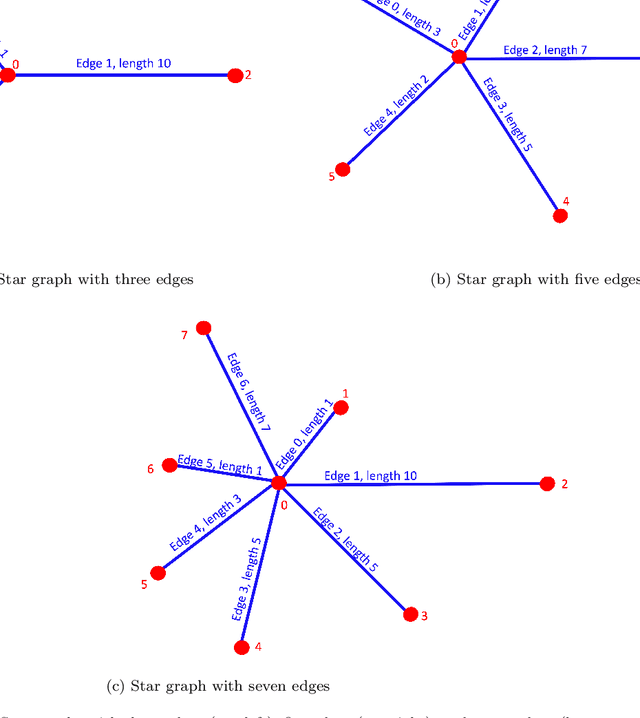
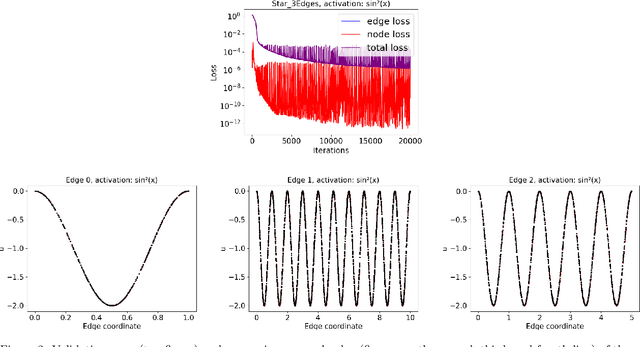
We propose a novel deep learning (DL) approach to solve one-dimensional non-linear elliptic, parabolic, and hyperbolic problems on graphs. A system of physics-informed neural network (PINN) models is used to solve the differential equations, by assigning each PINN model to a specific edge of the graph. Kirkhoff-Neumann (KN) nodal conditions are imposed in a weak form by adding a penalization term to the training loss function. Through the penalization term that imposes the KN conditions, PINN models associated with edges that share a node coordinate with each other to ensure continuity of the solution and of its directional derivatives computed along the respective edges. Using individual PINN models for each edge of the graph allows our approach to fulfill necessary requirements for parallelization by enabling different PINN models to be trained on distributed compute resources. Numerical results show that the system of PINN models accurately approximate the solutions of the differential problems across the entire graph for a broad set of graph topologies.
Stable Parallel Training of Wasserstein Conditional Generative Adversarial Neural Networks
Jul 25, 2022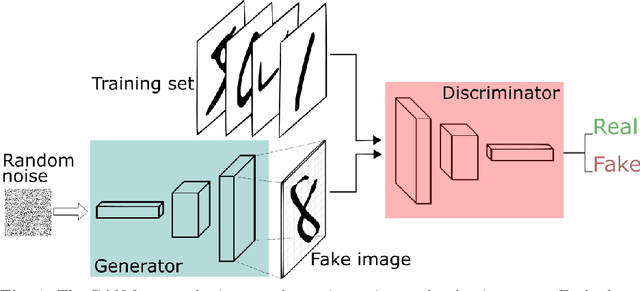
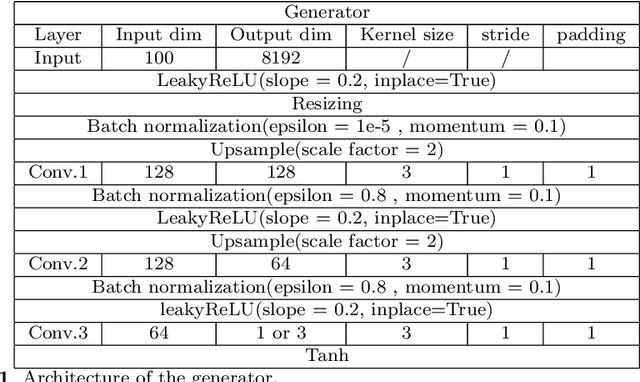
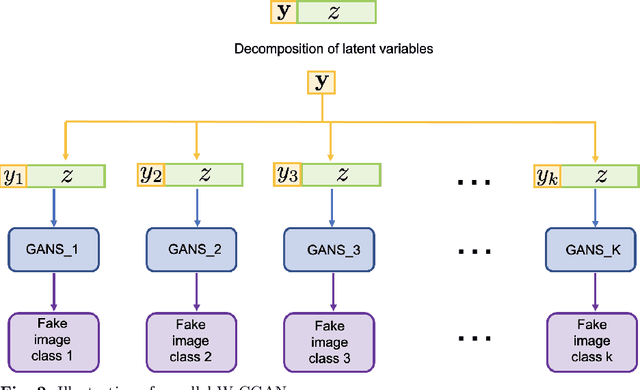
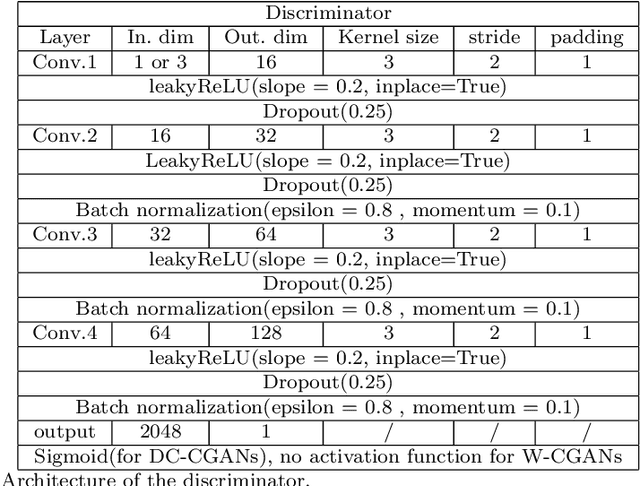
We propose a stable, parallel approach to train Wasserstein Conditional Generative Adversarial Neural Networks (W-CGANs) under the constraint of a fixed computational budget. Differently from previous distributed GANs training techniques, our approach avoids inter-process communications, reduces the risk of mode collapse and enhances scalability by using multiple generators, each one of them concurrently trained on a single data label. The use of the Wasserstein metric also reduces the risk of cycling by stabilizing the training of each generator. We illustrate the approach on the CIFAR10, CIFAR100, and ImageNet1k datasets, three standard benchmark image datasets, maintaining the original resolution of the images for each dataset. Performance is assessed in terms of scalability and final accuracy within a limited fixed computational time and computational resources. To measure accuracy, we use the inception score, the Frechet inception distance, and image quality. An improvement in inception score and Frechet inception distance is shown in comparison to previous results obtained by performing the parallel approach on deep convolutional conditional generative adversarial neural networks (DC-CGANs) as well as an improvement of image quality of the new images created by the GANs approach. Weak scaling is attained on both datasets using up to 2,000 NVIDIA V100 GPUs on the OLCF supercomputer Summit.
Scalable training of graph convolutional neural networks for fast and accurate predictions of HOMO-LUMO gap in molecules
Jul 22, 2022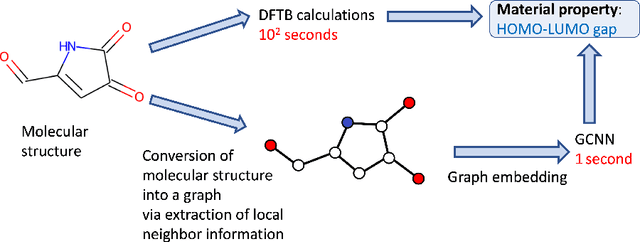

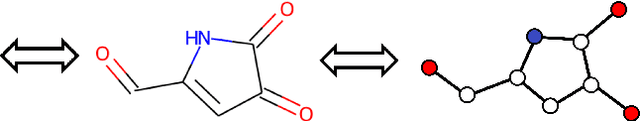
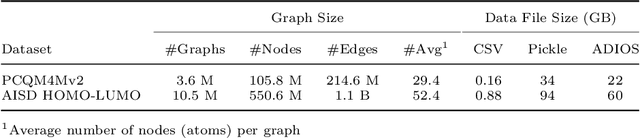
Graph Convolutional Neural Network (GCNN) is a popular class of deep learning (DL) models in material science to predict material properties from the graph representation of molecular structures. Training an accurate and comprehensive GCNN surrogate for molecular design requires large-scale graph datasets and is usually a time-consuming process. Recent advances in GPUs and distributed computing open a path to reduce the computational cost for GCNN training effectively. However, efficient utilization of high performance computing (HPC) resources for training requires simultaneously optimizing large-scale data management and scalable stochastic batched optimization techniques. In this work, we focus on building GCNN models on HPC systems to predict material properties of millions of molecules. We use HydraGNN, our in-house library for large-scale GCNN training, leveraging distributed data parallelism in PyTorch. We use ADIOS, a high-performance data management framework for efficient storage and reading of large molecular graph data. We perform parallel training on two open-source large-scale graph datasets to build a GCNN predictor for an important quantum property known as the HOMO-LUMO gap. We measure the scalability, accuracy, and convergence of our approach on two DOE supercomputers: the Summit supercomputer at the Oak Ridge Leadership Computing Facility (OLCF) and the Perlmutter system at the National Energy Research Scientific Computing Center (NERSC). We present our experimental results with HydraGNN showing i) reduction of data loading time up to 4.2 times compared with a conventional method and ii) linear scaling performance for training up to 1,024 GPUs on both Summit and Perlmutter.
Hierarchical model reduction driven by machine learning for parametric advection-diffusion-reaction problems in the presence of noisy data
Apr 01, 2022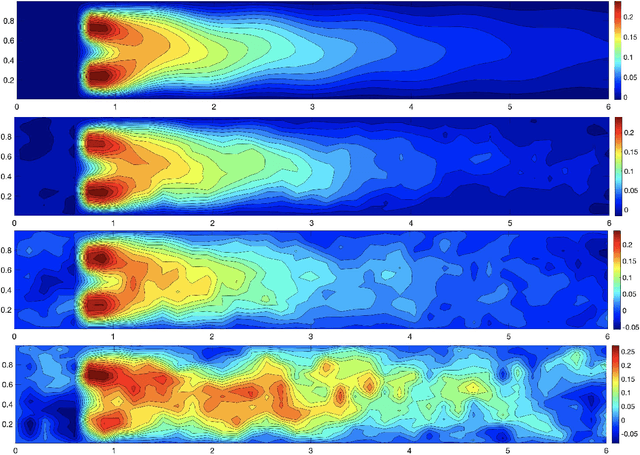
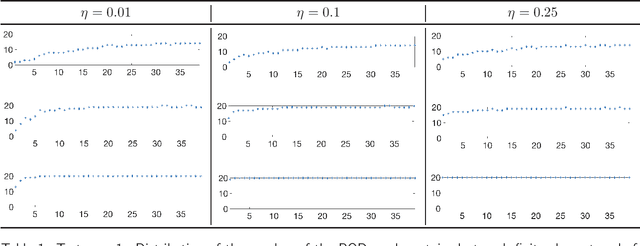
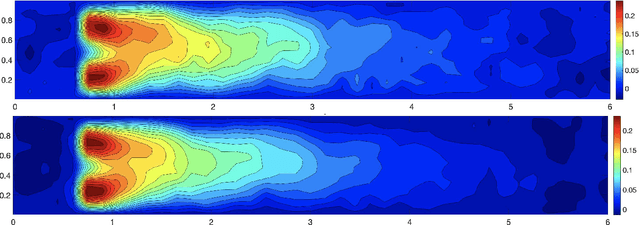
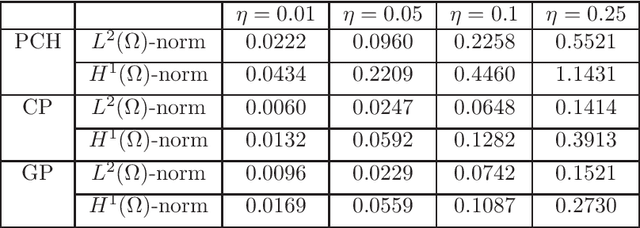
We propose a new approach to generate a reliable reduced model for a parametric elliptic problem, in the presence of noisy data. The reference model reduction procedure is the directional HiPOD method, which combines Hierarchical Model reduction with a standard Proper Orthogonal Decomposition, according to an offline/online paradigm. In this paper we show that directional HiPOD looses in terms of accuracy when problem data are affected by noise. This is due to the interpolation driving the online phase, since it replicates, by definition, the noise trend. To overcome this limit, we replace interpolation with Machine Learning fitting models which better discriminate relevant physical features in the data from irrelevant unstructured noise. The numerical assessment, although preliminary, confirms the potentialities of the new approach.