 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeQuantum-machine-assisted Drug Discovery: Survey and Perspective
Aug 24, 2024



Drug discovery and development is a highly complex and costly endeavor, typically requiring over a decade and substantial financial investment to bring a new drug to market. Traditional computer-aided drug design (CADD) has made significant progress in accelerating this process, but the development of quantum computing offers potential due to its unique capabilities. This paper discusses the integration of quantum computing into drug discovery and development, focusing on how quantum technologies might accelerate and enhance various stages of the drug development cycle. Specifically, we explore the application of quantum computing in addressing challenges related to drug discovery, such as molecular simulation and the prediction of drug-target interactions, as well as the optimization of clinical trial outcomes. By leveraging the inherent capabilities of quantum computing, we might be able to reduce the time and cost associated with bringing new drugs to market, ultimately benefiting public health.
MoExtend: Tuning New Experts for Modality and Task Extension
Aug 07, 2024



Large language models (LLMs) excel in various tasks but are primarily trained on text data, limiting their application scope. Expanding LLM capabilities to include vision-language understanding is vital, yet training them on multimodal data from scratch is challenging and costly. Existing instruction tuning methods, e.g., LLAVA, often connects a pretrained CLIP vision encoder and LLMs via fully fine-tuning LLMs to bridge the modality gap. However, full fine-tuning is plagued by catastrophic forgetting, i.e., forgetting previous knowledge, and high training costs particularly in the era of increasing tasks and modalities. To solve this issue, we introduce MoExtend, an effective framework designed to streamline the modality adaptation and extension of Mixture-of-Experts (MoE) models. MoExtend seamlessly integrates new experts into pre-trained MoE models, endowing them with novel knowledge without the need to tune pretrained models such as MoE and vision encoders. This approach enables rapid adaptation and extension to new modal data or tasks, effectively addressing the challenge of accommodating new modalities within LLMs. Furthermore, MoExtend avoids tuning pretrained models, thus mitigating the risk of catastrophic forgetting. Experimental results demonstrate the efficacy and efficiency of MoExtend in enhancing the multimodal capabilities of LLMs, contributing to advancements in multimodal AI research. Code: https://github.com/zhongshsh/MoExtend.
Composable Interventions for Language Models
Jul 09, 2024



Test-time interventions for language models can enhance factual accuracy, mitigate harmful outputs, and improve model efficiency without costly retraining. But despite a flood of new methods, different types of interventions are largely developing independently. In practice, multiple interventions must be applied sequentially to the same model, yet we lack standardized ways to study how interventions interact. We fill this gap by introducing composable interventions, a framework to study the effects of using multiple interventions on the same language models, featuring new metrics and a unified codebase. Using our framework, we conduct extensive experiments and compose popular methods from three emerging intervention categories -- Knowledge Editing, Model Compression, and Machine Unlearning. Our results from 310 different compositions uncover meaningful interactions: compression hinders editing and unlearning, composing interventions hinges on their order of application, and popular general-purpose metrics are inadequate for assessing composability. Taken together, our findings showcase clear gaps in composability, suggesting a need for new multi-objective interventions. All of our code is public: https://github.com/hartvigsen-group/composable-interventions.
TrialBench: Multi-Modal Artificial Intelligence-Ready Clinical Trial Datasets
Jun 30, 2024



Clinical trials are pivotal for developing new medical treatments, yet they typically pose some risks such as patient mortality, adverse events, and enrollment failure that waste immense efforts spanning over a decade. Applying artificial intelligence (AI) to forecast or simulate key events in clinical trials holds great potential for providing insights to guide trial designs. However, complex data collection and question definition requiring medical expertise and a deep understanding of trial designs have hindered the involvement of AI thus far. This paper tackles these challenges by presenting a comprehensive suite of meticulously curated AIready datasets covering multi-modal data (e.g., drug molecule, disease code, text, categorical/numerical features) and 8 crucial prediction challenges in clinical trial design, encompassing prediction of trial duration, patient dropout rate, serious adverse event, mortality rate, trial approval outcome, trial failure reason, drug dose finding, design of eligibility criteria. Furthermore, we provide basic validation methods for each task to ensure the datasets' usability and reliability. We anticipate that the availability of such open-access datasets will catalyze the development of advanced AI approaches for clinical trial design, ultimately advancing clinical trial research and accelerating medical solution development. The curated dataset, metrics, and basic models are publicly available at https://github.com/ML2Health/ML2ClinicalTrials/tree/main/AI4Trial.
Graph Adversarial Diffusion Convolution
Jun 04, 2024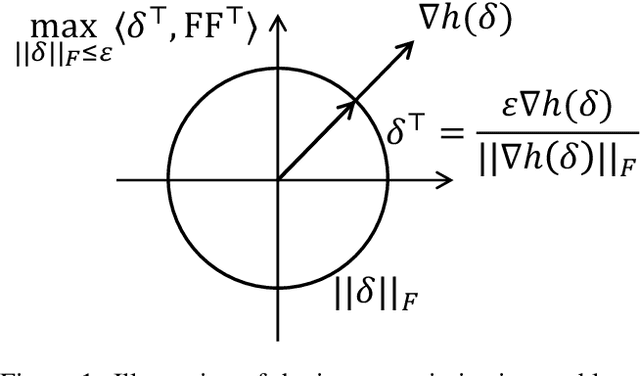
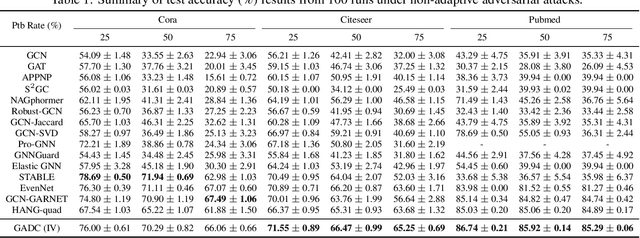
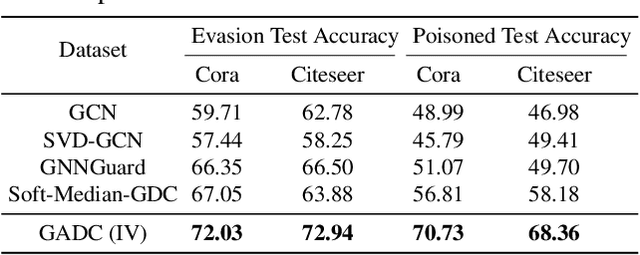
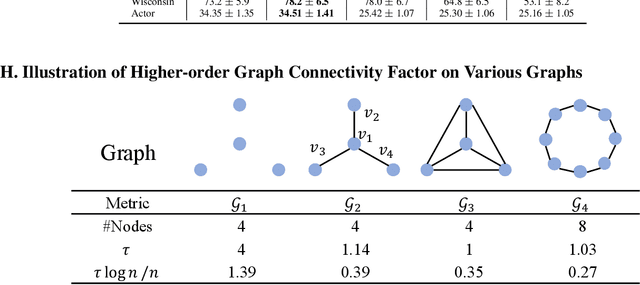
This paper introduces a min-max optimization formulation for the Graph Signal Denoising (GSD) problem. In this formulation, we first maximize the second term of GSD by introducing perturbations to the graph structure based on Laplacian distance and then minimize the overall loss of the GSD. By solving the min-max optimization problem, we derive a new variant of the Graph Diffusion Convolution (GDC) architecture, called Graph Adversarial Diffusion Convolution (GADC). GADC differs from GDC by incorporating an additional term that enhances robustness against adversarial attacks on the graph structure and noise in node features. Moreover, GADC improves the performance of GDC on heterophilic graphs. Extensive experiments demonstrate the effectiveness of GADC across various datasets. Code is available at https://github.com/SongtaoLiu0823/GADC.
Structure-based Drug Design Benchmark: Do 3D Methods Really Dominate?
Jun 04, 2024Currently, the field of structure-based drug design is dominated by three main types of algorithms: search-based algorithms, deep generative models, and reinforcement learning. While existing works have typically focused on comparing models within a single algorithmic category, cross-algorithm comparisons remain scarce. In this paper, to fill the gap, we establish a benchmark to evaluate the performance of sixteen models across these different algorithmic foundations by assessing the pharmaceutical properties of the generated molecules and their docking affinities with specified target proteins. We highlight the unique advantages of each algorithmic approach and offer recommendations for the design of future SBDD models. We emphasize that 1D/2D ligand-centric drug design methods can be used in SBDD by treating the docking function as a black-box oracle, which is typically neglected. The empirical results show that 1D/2D methods achieve competitive performance compared with 3D-based methods that use the 3D structure of the target protein explicitly. Also, AutoGrow4, a 2D molecular graph-based genetic algorithm, dominates SBDD in terms of optimization ability. The relevant code is available in https://github.com/zkysfls/2024-sbdd-benchmark.
Learning Generalized Medical Image Representations through Image-Graph Contrastive Pretraining
May 15, 2024



Medical image interpretation using deep learning has shown promise but often requires extensive expert-annotated datasets. To reduce this annotation burden, we develop an Image-Graph Contrastive Learning framework that pairs chest X-rays with structured report knowledge graphs automatically extracted from radiology notes. Our approach uniquely encodes the disconnected graph components via a relational graph convolution network and transformer attention. In experiments on the CheXpert dataset, this novel graph encoding strategy enabled the framework to outperform existing methods that use image-text contrastive learning in 1% linear evaluation and few-shot settings, while achieving comparable performance to radiologists. By exploiting unlabeled paired images and text, our framework demonstrates the potential of structured clinical insights to enhance contrastive learning for medical images. This work points toward reducing demands on medical experts for annotations, improving diagnostic precision, and advancing patient care through robust medical image understanding.
Empowering Biomedical Discovery with AI Agents
Apr 03, 2024



We envision 'AI scientists' as systems capable of skeptical learning and reasoning that empower biomedical research through collaborative agents that integrate machine learning tools with experimental platforms. Rather than taking humans out of the discovery process, biomedical AI agents combine human creativity and expertise with AI's ability to analyze large datasets, navigate hypothesis spaces, and execute repetitive tasks. AI agents are proficient in a variety of tasks, including self-assessment and planning of discovery workflows. These agents use large language models and generative models to feature structured memory for continual learning and use machine learning tools to incorporate scientific knowledge, biological principles, and theories. AI agents can impact areas ranging from hybrid cell simulation, programmable control of phenotypes, and the design of cellular circuits to the development of new therapies.
Recent Advances, Applications, and Open Challenges in Machine Learning for Health: Reflections from Research Roundtables at ML4H 2023 Symposium
Mar 03, 2024The third ML4H symposium was held in person on December 10, 2023, in New Orleans, Louisiana, USA. The symposium included research roundtable sessions to foster discussions between participants and senior researchers on timely and relevant topics for the \ac{ML4H} community. Encouraged by the successful virtual roundtables in the previous year, we organized eleven in-person roundtables and four virtual roundtables at ML4H 2022. The organization of the research roundtables at the conference involved 17 Senior Chairs and 19 Junior Chairs across 11 tables. Each roundtable session included invited senior chairs (with substantial experience in the field), junior chairs (responsible for facilitating the discussion), and attendees from diverse backgrounds with interest in the session's topic. Herein we detail the organization process and compile takeaways from these roundtable discussions, including recent advances, applications, and open challenges for each topic. We conclude with a summary and lessons learned across all roundtables. This document serves as a comprehensive review paper, summarizing the recent advancements in machine learning for healthcare as contributed by foremost researchers in the field.
UniTS: Building a Unified Time Series Model
Feb 29, 2024



Foundation models, especially LLMs, are profoundly transforming deep learning. Instead of training many task-specific models, we can adapt a single pretrained model to many tasks via fewshot prompting or fine-tuning. However, current foundation models apply to sequence data but not to time series, which present unique challenges due to the inherent diverse and multidomain time series datasets, diverging task specifications across forecasting, classification and other types of tasks, and the apparent need for task-specialized models. We developed UNITS, a unified time series model that supports a universal task specification, accommodating classification, forecasting, imputation, and anomaly detection tasks. This is achieved through a novel unified network backbone, which incorporates sequence and variable attention along with a dynamic linear operator and is trained as a unified model. Across 38 multi-domain datasets, UNITS demonstrates superior performance compared to task-specific models and repurposed natural language-based LLMs. UNITS exhibits remarkable zero-shot, few-shot, and prompt learning capabilities when evaluated on new data domains and tasks. The source code and datasets are available at https://github.com/mims-harvard/UniTS.