 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAI Competitions and Benchmarks: The life cycle of challenges and benchmarks
Dec 08, 2023

Data Science research is undergoing a revolution fueled by the transformative power of technology, the Internet, and an ever increasing computational capacity. The rate at which sophisticated algorithms can be developed is unprecedented, yet they remain outpaced by the massive amounts of data that are increasingly available to researchers. Here we argue for the need to creatively leverage the scientific research and algorithm development community as an axis of robust innovation. Engaging these communities in the scientific discovery enterprise by critical assessments, community experiments, and/or crowdsourcing will multiply opportunities to develop new data driven, reproducible and well benchmarked algorithmic solutions to fundamental and applied problems of current interest. Coordinated community engagement in the analysis of highly complex and massive data has emerged as one approach to find robust methodologies that best address these challenges. When community engagement is done in the form of competitions, also known as challenges, the validation of the analytical methodology is inherently addressed, establishing performance benchmarks. Finally, challenges foster open innovation across multiple disciplines to create communities that collaborate directly or indirectly to address significant scientific gaps. Together, participants can solve important problems as varied as health research, climate change, and social equity. Ultimately, challenges can catalyze and accelerate the synthesis of complex data into knowledge or actionable information, and should be viewed a powerful tool to make lasting social and research contributions.
Understanding metric-related pitfalls in image analysis validation
Feb 09, 2023Validation metrics are key for the reliable tracking of scientific progress and for bridging the current chasm between artificial intelligence (AI) research and its translation into practice. However, increasing evidence shows that particularly in image analysis, metrics are often chosen inadequately in relation to the underlying research problem. This could be attributed to a lack of accessibility of metric-related knowledge: While taking into account the individual strengths, weaknesses, and limitations of validation metrics is a critical prerequisite to making educated choices, the relevant knowledge is currently scattered and poorly accessible to individual researchers. Based on a multi-stage Delphi process conducted by a multidisciplinary expert consortium as well as extensive community feedback, the present work provides the first reliable and comprehensive common point of access to information on pitfalls related to validation metrics in image analysis. Focusing on biomedical image analysis but with the potential of transfer to other fields, the addressed pitfalls generalize across application domains and are categorized according to a newly created, domain-agnostic taxonomy. To facilitate comprehension, illustrations and specific examples accompany each pitfall. As a structured body of information accessible to researchers of all levels of expertise, this work enhances global comprehension of a key topic in image analysis validation.
Metrics reloaded: Pitfalls and recommendations for image analysis validation
Jun 03, 2022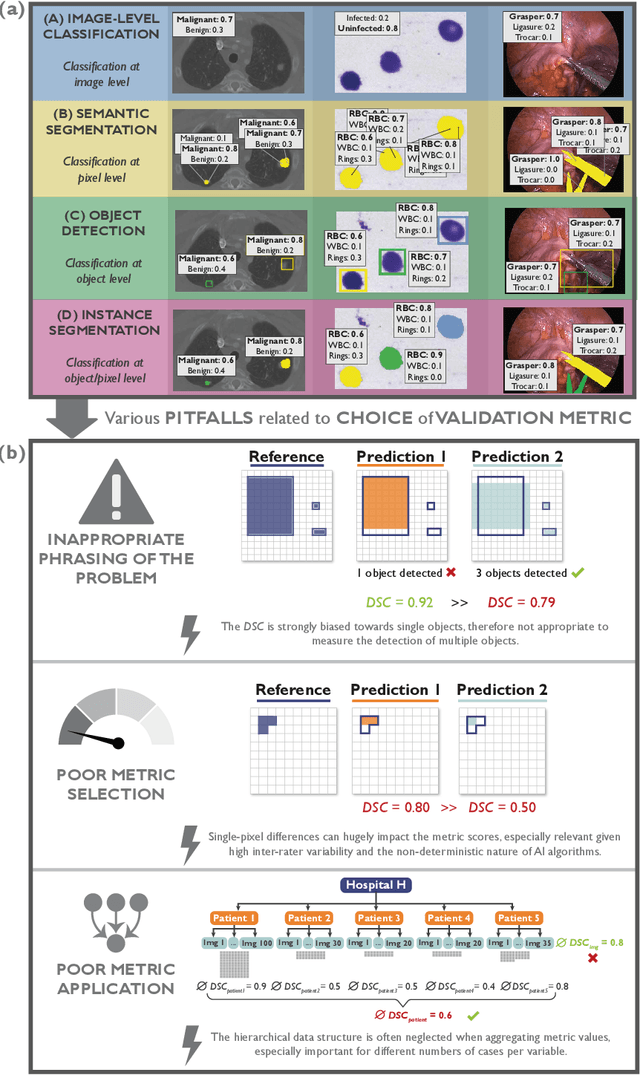
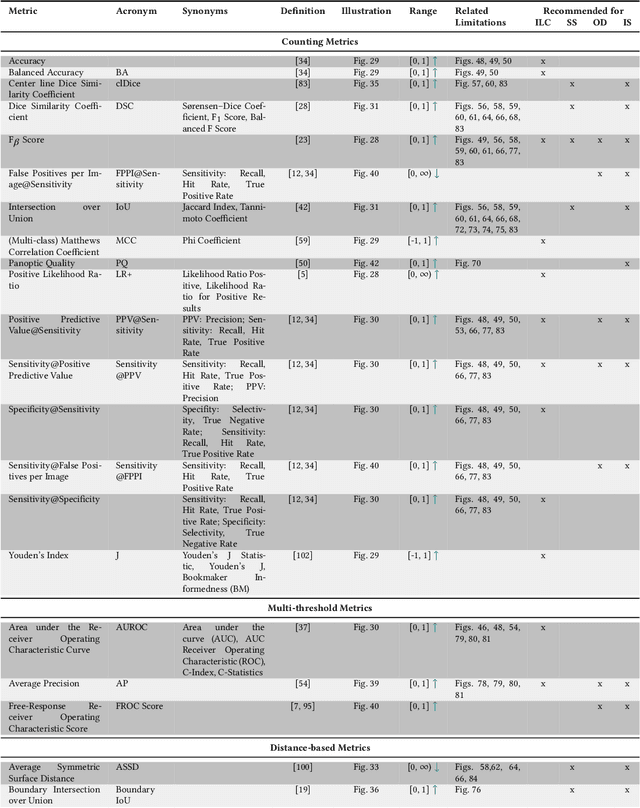
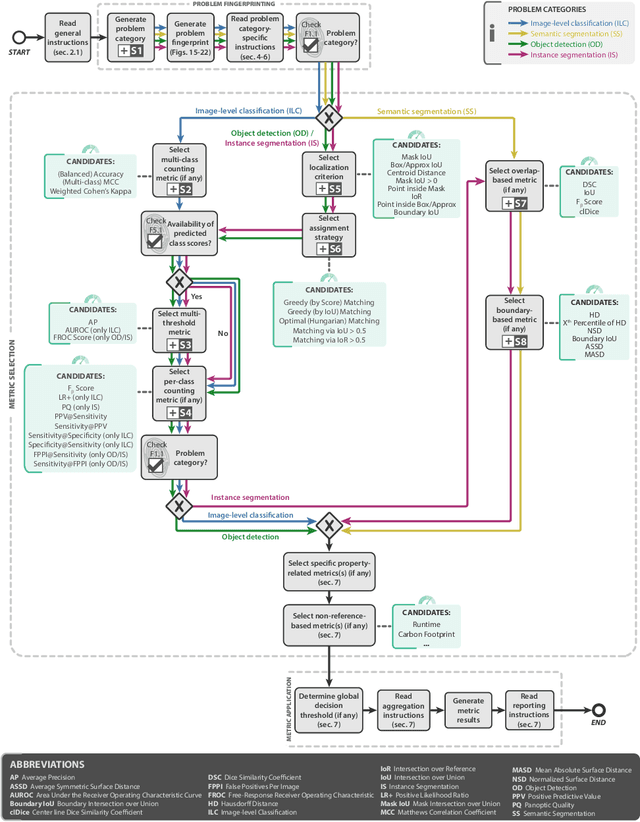
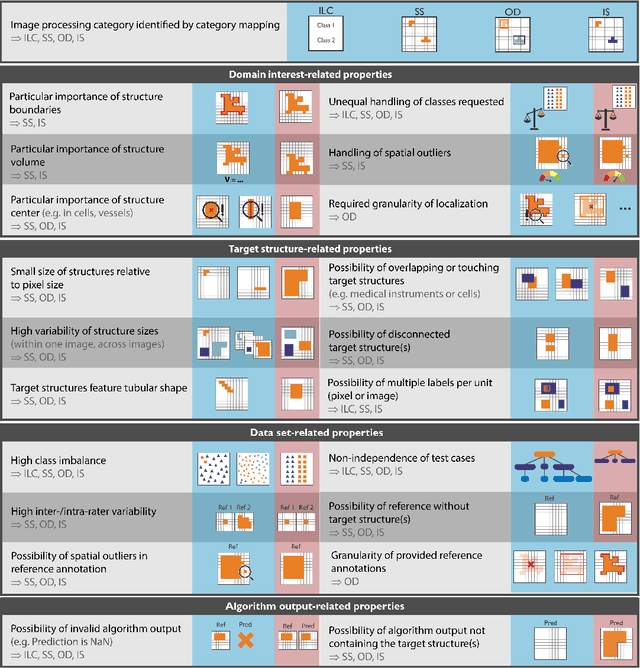
The field of automatic biomedical image analysis crucially depends on robust and meaningful performance metrics for algorithm validation. Current metric usage, however, is often ill-informed and does not reflect the underlying domain interest. Here, we present a comprehensive framework that guides researchers towards choosing performance metrics in a problem-aware manner. Specifically, we focus on biomedical image analysis problems that can be interpreted as a classification task at image, object or pixel level. The framework first compiles domain interest-, target structure-, data set- and algorithm output-related properties of a given problem into a problem fingerprint, while also mapping it to the appropriate problem category, namely image-level classification, semantic segmentation, instance segmentation, or object detection. It then guides users through the process of selecting and applying a set of appropriate validation metrics while making them aware of potential pitfalls related to individual choices. In this paper, we describe the current status of the Metrics Reloaded recommendation framework, with the goal of obtaining constructive feedback from the image analysis community. The current version has been developed within an international consortium of more than 60 image analysis experts and will be made openly available as a user-friendly toolkit after community-driven optimization.
BIAS: Transparent reporting of biomedical image analysis challenges
Oct 23, 2019The number of biomedical image analysis challenges organized per year is steadily increasing. These international competitions have the purpose of benchmarking algorithms on common data sets, typically to identify the best method for a given problem. Recent research, however, revealed that common practice related to challenge reporting does not allow for adequate interpretation and reproducibility of results. To address the discrepancy between the impact of challenges and the quality (control), the Biomedical I mage Analysis ChallengeS (BIAS) initiative developed a set of recommendations for the reporting of challenges. The BIAS statement aims to improve the transparency of the reporting of a biomedical image analysis challenge regardless of field of application, image modality or task category assessed. This article describes how the BIAS statement was developed and presents a checklist which authors of biomedical image analysis challenges are encouraged to include in their submission when giving a paper on a challenge into review. The purpose of the checklist is to standardize and facilitate the review process and raise interpretability and reproducibility of challenge results by making relevant information explicit.
Machine learning prediction of cancer cell sensitivity to drugs based on genomic and chemical properties
Mar 18, 2013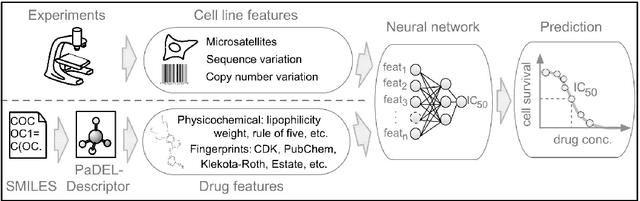
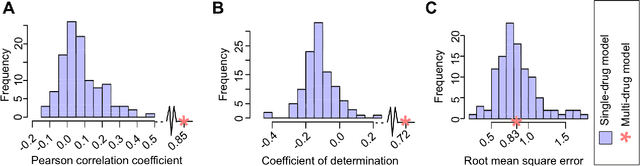
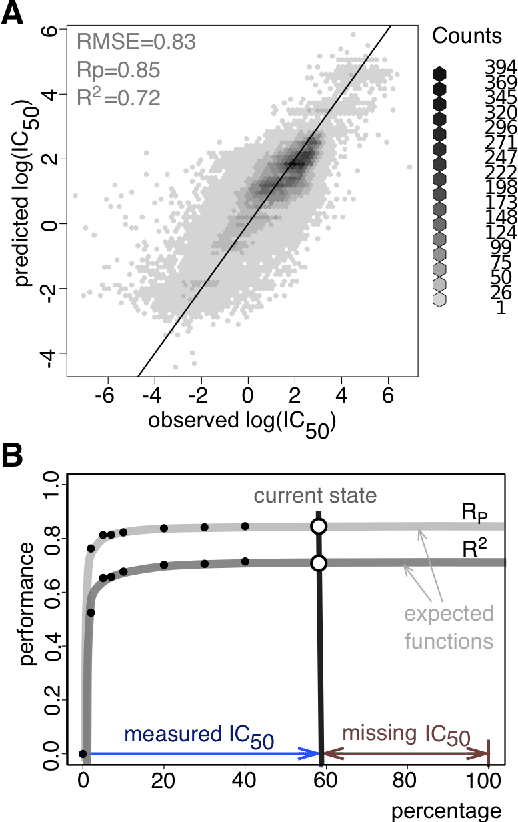
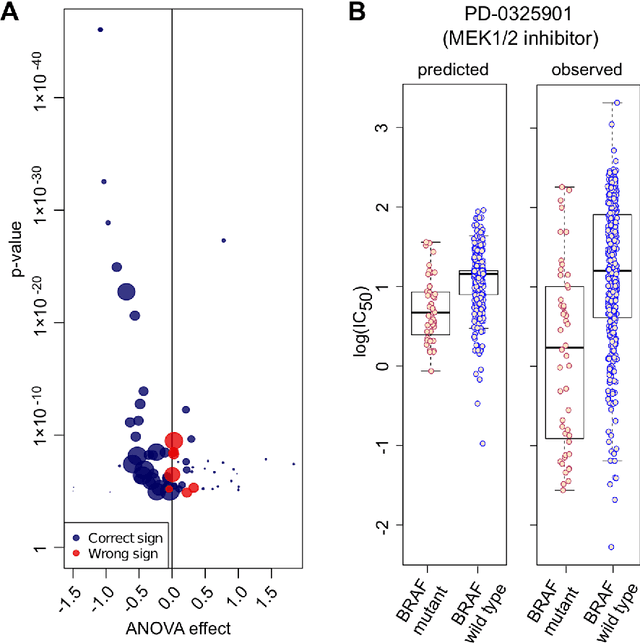
Predicting the response of a specific cancer to a therapy is a major goal in modern oncology that should ultimately lead to a personalised treatment. High-throughput screenings of potentially active compounds against a panel of genomically heterogeneous cancer cell lines have unveiled multiple relationships between genomic alterations and drug responses. Various computational approaches have been proposed to predict sensitivity based on genomic features, while others have used the chemical properties of the drugs to ascertain their effect. In an effort to integrate these complementary approaches, we developed machine learning models to predict the response of cancer cell lines to drug treatment, quantified through IC50 values, based on both the genomic features of the cell lines and the chemical properties of the considered drugs. Models predicted IC50 values in a 8-fold cross-validation and an independent blind test with coefficient of determination R2 of 0.72 and 0.64 respectively. Furthermore, models were able to predict with comparable accuracy (R2 of 0.61) IC50s of cell lines from a tissue not used in the training stage. Our in silico models can be used to optimise the experimental design of drug-cell screenings by estimating a large proportion of missing IC50 values rather than experimentally measure them. The implications of our results go beyond virtual drug screening design: potentially thousands of drugs could be probed in silico to systematically test their potential efficacy as anti-tumour agents based on their structure, thus providing a computational framework to identify new drug repositioning opportunities as well as ultimately be useful for personalized medicine by linking the genomic traits of patients to drug sensitivity.
Revisiting the Training of Logic Models of Protein Signaling Networks with a Formal Approach based on Answer Set Programming
Dec 22, 2012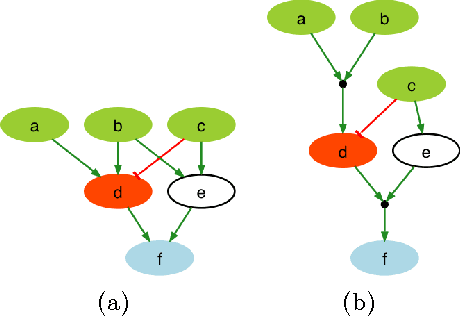
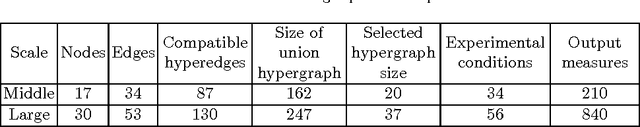
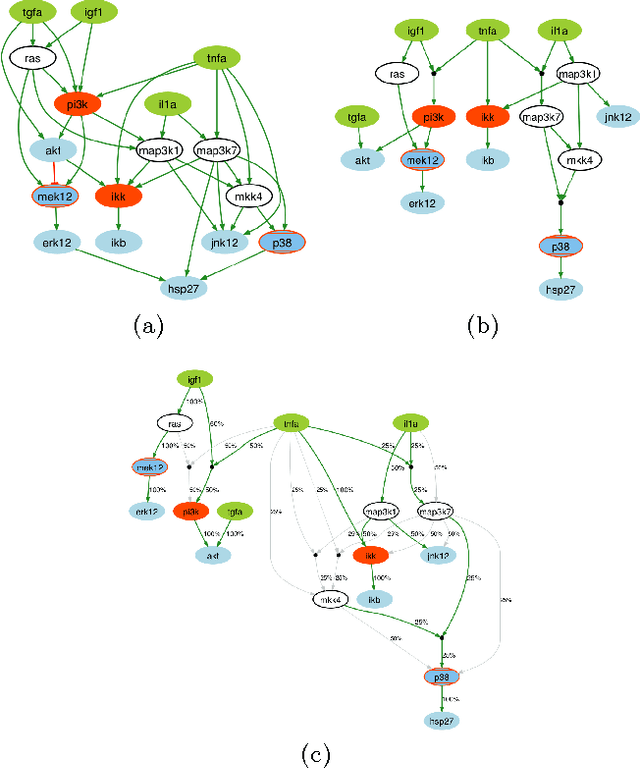
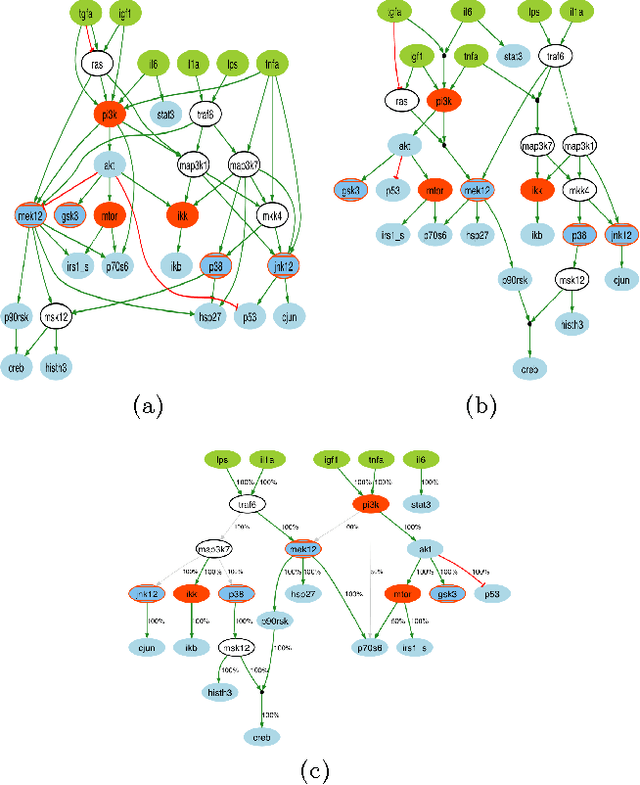
A fundamental question in systems biology is the construction and training to data of mathematical models. Logic formalisms have become very popular to model signaling networks because their simplicity allows us to model large systems encompassing hundreds of proteins. An approach to train (Boolean) logic models to high-throughput phospho-proteomics data was recently introduced and solved using optimization heuristics based on stochastic methods. Here we demonstrate how this problem can be solved using Answer Set Programming (ASP), a declarative problem solving paradigm, in which a problem is encoded as a logical program such that its answer sets represent solutions to the problem. ASP has significant improvements over heuristic methods in terms of efficiency and scalability, it guarantees global optimality of solutions as well as provides a complete set of solutions. We illustrate the application of ASP with in silico cases based on realistic networks and data.