 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeArtificial Intelligence for Science in Quantum, Atomistic, and Continuum Systems
Jul 17, 2023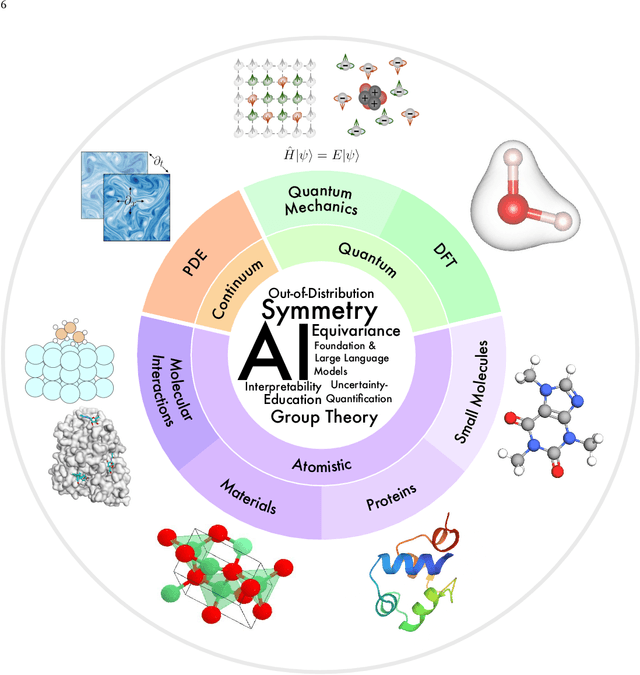



Advances in artificial intelligence (AI) are fueling a new paradigm of discoveries in natural sciences. Today, AI has started to advance natural sciences by improving, accelerating, and enabling our understanding of natural phenomena at a wide range of spatial and temporal scales, giving rise to a new area of research known as AI for science (AI4Science). Being an emerging research paradigm, AI4Science is unique in that it is an enormous and highly interdisciplinary area. Thus, a unified and technical treatment of this field is needed yet challenging. This paper aims to provide a technically thorough account of a subarea of AI4Science; namely, AI for quantum, atomistic, and continuum systems. These areas aim at understanding the physical world from the subatomic (wavefunctions and electron density), atomic (molecules, proteins, materials, and interactions), to macro (fluids, climate, and subsurface) scales and form an important subarea of AI4Science. A unique advantage of focusing on these areas is that they largely share a common set of challenges, thereby allowing a unified and foundational treatment. A key common challenge is how to capture physics first principles, especially symmetries, in natural systems by deep learning methods. We provide an in-depth yet intuitive account of techniques to achieve equivariance to symmetry transformations. We also discuss other common technical challenges, including explainability, out-of-distribution generalization, knowledge transfer with foundation and large language models, and uncertainty quantification. To facilitate learning and education, we provide categorized lists of resources that we found to be useful. We strive to be thorough and unified and hope this initial effort may trigger more community interests and efforts to further advance AI4Science.
PyTrial: A Comprehensive Platform for Artificial Intelligence for Drug Development
Jun 06, 2023



Drug development is a complex process that aims to test the efficacy and safety of candidate drugs in the human body for regulatory approval via clinical trials. Recently, machine learning has emerged as a vital tool for drug development, offering new opportunities to improve the efficiency and success rates of the process. To facilitate the research and development of artificial intelligence (AI) for drug development, we developed a Python package, namely PyTrial, that implements various clinical trial tasks supported by AI algorithms. To be specific, PyTrial implements 6 essential drug development tasks, including patient outcome prediction, trial site selection, trial outcome prediction, patient-trial matching, trial similarity search, and synthetic data generation. In PyTrial, all tasks are defined by four steps: load data, model definition, model training, and model evaluation, which can be done with a couple of lines of code. In addition, the modular API design allows practitioners to extend the framework to new algorithms and tasks easily. PyTrial is featured for a unified API, detailed documentation, and interactive examples with preprocessed benchmark data for all implemented algorithms. This package can be installed through Python Package Index (PyPI) and is publicly available at https://github.com/RyanWangZf/PyTrial.
Generating with Confidence: Uncertainty Quantification for Black-box Large Language Models
May 30, 2023



Large language models (LLMs) specializing in natural language generation (NLG) have recently started exhibiting promising capabilities across a variety of domains. However, gauging the trustworthiness of responses generated by LLMs remains an open challenge, with limited research on uncertainty quantification for NLG. Furthermore, existing literature typically assumes white-box access to language models, which is becoming unrealistic either due to the closed-source nature of the latest LLMs or due to computational constraints. In this work, we investigate uncertainty quantification in NLG for $\textit{black-box}$ LLMs. We first differentiate two closely-related notions: $\textit{uncertainty}$, which depends only on the input, and $\textit{confidence}$, which additionally depends on the generated response. We then propose and compare several confidence/uncertainty metrics, applying them to $\textit{selective NLG}$, where unreliable results could either be ignored or yielded for further assessment. Our findings on several popular LLMs and datasets reveal that a simple yet effective metric for the average semantic dispersion can be a reliable predictor of the quality of LLM responses. This study can provide valuable insights for practitioners on uncertainty management when adopting LLMs. The code to replicate all our experiments is available at https://github.com/zlin7/UQ-NLG.
FRAMM: Fair Ranking with Missing Modalities for Clinical Trial Site Selection
May 30, 2023Despite many efforts to address the disparities, the underrepresentation of gender, racial, and ethnic minorities in clinical trials remains a problem and undermines the efficacy of treatments on minorities. This paper focuses on the trial site selection task and proposes FRAMM, a deep reinforcement learning framework for fair trial site selection. We focus on addressing two real-world challenges that affect fair trial sites selection: the data modalities are often not complete for many potential trial sites, and the site selection needs to simultaneously optimize for both enrollment and diversity since the problem is necessarily a trade-off between the two with the only possible way to increase diversity post-selection being through limiting enrollment via caps. To address the missing data challenge, FRAMM has a modality encoder with a masked cross-attention mechanism for handling missing data, bypassing data imputation and the need for complete data in training. To handle the need for making efficient trade-offs, FRAMM uses deep reinforcement learning with a specifically designed reward function that simultaneously optimizes for both enrollment and fairness. We evaluate FRAMM using 4,392 real-world clinical trials ranging from 2016 to 2021 and show that FRAMM outperforms the leading baseline in enrollment-only settings while also achieving large gains in diversity. Specifically, it is able to produce a 9% improvement in diversity with similar enrollment levels over the leading baselines. That improved diversity is further manifested in achieving up to a 14% increase in Hispanic enrollment, 27% increase in Black enrollment, and 60% increase in Asian enrollment compared to selecting sites with an enrollment-only model.
Text-Augmented Open Knowledge Graph Completion via Pre-Trained Language Models
May 24, 2023



The mission of open knowledge graph (KG) completion is to draw new findings from known facts. Existing works that augment KG completion require either (1) factual triples to enlarge the graph reasoning space or (2) manually designed prompts to extract knowledge from a pre-trained language model (PLM), exhibiting limited performance and requiring expensive efforts from experts. To this end, we propose TAGREAL that automatically generates quality query prompts and retrieves support information from large text corpora to probe knowledge from PLM for KG completion. The results show that TAGREAL achieves state-of-the-art performance on two benchmark datasets. We find that TAGREAL has superb performance even with limited training data, outperforming existing embedding-based, graph-based, and PLM-based methods.
GraphCare: Enhancing Healthcare Predictions with Open-World Personalized Knowledge Graphs
May 22, 2023Clinical predictive models often rely on patients electronic health records (EHR), but integrating medical knowledge to enhance predictions and decision-making is challenging. This is because personalized predictions require personalized knowledge graphs (KGs), which are difficult to generate from patient EHR data. To address this, we propose GraphCare, an open-world framework that leverages external KGs to improve EHR-based predictions. Our method extracts knowledge from large language models (LLMs) and external biomedical KGs to generate patient-specific KGs, which are then used to train our proposed Bi-attention AugmenTed BAT graph neural network GNN for healthcare predictions. We evaluate GraphCare on two public datasets: MIMIC-III and MIMIC-IV. Our method outperforms baseline models in four vital healthcare prediction tasks: mortality, readmission, length-of-stay, and drug recommendation, improving AUROC on MIMIC-III by average margins of 10.4%, 3.8%, 2.0%, and 1.5%, respectively. Notably, GraphCare demonstrates a substantial edge in scenarios with limited data availability. Our findings highlight the potential of using external KGs in healthcare prediction tasks and demonstrate the promise of GraphCare in generating personalized KGs for promoting personalized medicine.
AnyPredict: Foundation Model for Tabular Prediction
May 20, 2023Foundation models are pre-trained on massive data to perform well across many downstream tasks. They have demonstrated significant success in natural language processing and computer vision. Nonetheless, the use of such models in tabular prediction tasks has been limited, with the main hurdles consisting of (1) the lack of large-scale and diverse tabular datasets with standardized labels and (2) the schema mismatch and predictive target heterogeneity across domains. This paper proposes a method for building training data at scale for tabular prediction foundation models (AnyPredict) using both in-domain and a wide range of out-domain datasets. The method uses a data engine that leverages large language models (LLMs) to consolidate tabular samples to overcome the barrier across tables with varying schema and align out-domain data with the target task using a ``learn, annotate, and audit'' pipeline. The expanded training data enables the pre-trained AnyPredict to support every tabular dataset in the domain without fine-tuning, resulting in significant improvements over supervised baselines: it reaches an average ranking of 1.57 and 1.00 on 7 patient outcome prediction datasets and 3 trial outcome prediction datasets, respectively. In addition, AnyPredict exhibits impressive zero-shot performances: it outperforms supervised XGBoost models by 8.9% and 17.2% on average in two prediction tasks, respectively.
AutoTrial: Prompting Language Models for Clinical Trial Design
May 19, 2023Clinical trials are critical for drug development. Constructing the appropriate eligibility criteria (i.e., the inclusion/exclusion criteria for patient recruitment) is essential for the trial's success. Proper design of clinical trial protocols should consider similar precedent trials and their eligibility criteria to ensure sufficient patient coverage. In this paper, we present a method named AutoTrial to aid the design of clinical eligibility criteria using language models. It allows (1) controllable generation under instructions via a hybrid of discrete and neural prompting, (2) scalable knowledge incorporation via in-context learning, and (3) explicit reasoning chains to provide rationales for understanding the outputs. Experiments on over 70K clinical trials verify that AutoTrial generates high-quality criteria texts that are fluent and coherent and with high accuracy in capturing the relevant clinical concepts to the target trial. It is noteworthy that our method, with a much smaller parameter size, gains around 60\% winning rate against the GPT-3.5 baselines via human evaluations.
BIOT: Cross-data Biosignal Learning in the Wild
May 10, 2023Biological signals, such as electroencephalograms (EEG), play a crucial role in numerous clinical applications, exhibiting diverse data formats and quality profiles. Current deep learning models for biosignals are typically specialized for specific datasets and clinical settings, limiting their broader applicability. Motivated by the success of large language models in text processing, we explore the development of foundational models that are trained from multiple data sources and can be fine-tuned on different downstream biosignal tasks. To overcome the unique challenges associated with biosignals of various formats, such as mismatched channels, variable sample lengths, and prevalent missing values, we propose a Biosignal Transformer (\method). The proposed \method model can enable cross-data learning with mismatched channels, variable lengths, and missing values by tokenizing diverse biosignals into unified "biosignal sentences". Specifically, we tokenize each channel into fixed-length segments containing local signal features, flattening them to form consistent "sentences". Channel embeddings and {\em relative} position embeddings are added to preserve spatio-temporal features. The \method model is versatile and applicable to various biosignal learning settings across different datasets, including joint pre-training for larger models. Comprehensive evaluations on EEG, electrocardiogram (ECG), and human activity sensory signals demonstrate that \method outperforms robust baselines in common settings and facilitates learning across multiple datasets with different formats. Use CHB-MIT seizure detection task as an example, our vanilla \method model shows 3\% improvement over baselines in balanced accuracy, and the pre-trained \method models (optimized from other data sources) can further bring up to 4\% improvements.
SPOT: Sequential Predictive Modeling of Clinical Trial Outcome with Meta-Learning
Apr 07, 2023Clinical trials are essential to drug development but time-consuming, costly, and prone to failure. Accurate trial outcome prediction based on historical trial data promises better trial investment decisions and more trial success. Existing trial outcome prediction models were not designed to model the relations among similar trials, capture the progression of features and designs of similar trials, or address the skewness of trial data which causes inferior performance for less common trials. To fill the gap and provide accurate trial outcome prediction, we propose Sequential Predictive mOdeling of clinical Trial outcome (SPOT) that first identifies trial topics to cluster the multi-sourced trial data into relevant trial topics. It then generates trial embeddings and organizes them by topic and time to create clinical trial sequences. With the consideration of each trial sequence as a task, it uses a meta-learning strategy to achieve a point where the model can rapidly adapt to new tasks with minimal updates. In particular, the topic discovery module enables a deeper understanding of the underlying structure of the data, while sequential learning captures the evolution of trial designs and outcomes. This results in predictions that are not only more accurate but also more interpretable, taking into account the temporal patterns and unique characteristics of each trial topic. We demonstrate that SPOT wins over the prior methods by a significant margin on trial outcome benchmark data: with a 21.5\% lift on phase I, an 8.9\% lift on phase II, and a 5.5\% lift on phase III trials in the metric of the area under precision-recall curve (PR-AUC).