 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAlloGen: Conformation-Selective Binder Generation with Differential State Scoring
Jun 03, 2026Protein binder design has largely optimized for affinity alone, leaving conformational selectivity unaddressed: for allosteric targets such as kinases, nuclear receptors, and GPCRs, a binder that engages both active and inactive states provides no functional specificity regardless of how tightly it binds. We introduce AlloGen, a modular framework that decouples backbone generation from a learned state-selectivity scorer $Q_θ$, an SE(3)-invariant interface graph transformer trained via a two-phase curriculum that first learns interface geometry before imposing conformational discrimination. Because $Q_θ$ is fully differentiable and generator-agnostic, it integrates with any backbone generator as a passive reranker or an active gradient-based guide without retraining. Across a diverse benchmark of proteins spanning multiple families and conformational mechanisms, AlloGen consistently identifies binders that preferentially recognize desired structural states while rejecting alternative conformations. Experimental validation on calmodulin further demonstrates that these computational selectivity signals translate to physical molecules, yielding de novo peptides that bind the desired holo conformation while exhibiting no detectable binding to the apo state. Together, these results establish conformational selectivity as a learnable property and provide a general framework for state-selective protein binder design.
TD3B: Transition-Directed Discrete Diffusion for Allosteric Binder Generation
May 10, 2026Protein function is often controlled by ligands that bias the direction of state transitions, such as agonists and antagonists, rather than stabilizing a single conformation. This is especially important for clinically relevant G protein-coupled receptors (GPCRs), where therapeutic efficacy depends on functional directionality. Structure-based design methods optimize binding to static conformations and cannot represent non-reversible, directional effects or systematically distinguish agonist from antagonist behavior. To address this gap, we introduce Transition-Directed Discrete Diffusion for Allosteric Binder Design (TD3B), a sequence-based generative framework that designs binders with specified agonist or antagonist behavior via a directional transition control objective. TD3B combines a target-aware Direction Oracle, a soft binding-affinity gate, and amortized fine-tuning of a pre-trained discrete diffusion model, enabling targeted agonist and antagonist generation decoupled from binding affinity and unattainable by equilibrium-based or inference-only guidance baselines. The code and checkpoints are available at https://huggingface.co/ChatterjeeLab/TD3B.
RiboSphere: Learning Unified and Efficient Representations of RNA Structures
Mar 20, 2026Accurate RNA structure modeling remains difficult because RNA backbones are highly flexible, non-canonical interactions are prevalent, and experimentally determined 3D structures are comparatively scarce. We introduce \emph{RiboSphere}, a framework that learns \emph{discrete} geometric representations of RNA by combining vector quantization with flow matching. Our design is motivated by the modular organization of RNA architecture: complex folds are composed from recurring structural motifs. RiboSphere uses a geometric transformer encoder to produce SE(3)-invariant (rotation/translation-invariant) features, which are discretized with finite scalar quantization (FSQ) into a finite vocabulary of latent codes. Conditioned on these discrete codes, a flow-matching decoder reconstructs atomic coordinates, enabling high-fidelity structure generation. We find that the learned code indices are enriched for specific RNA motifs, suggesting that the model captures motif-level compositional structure rather than acting as a purely compressive bottleneck. Across benchmarks, RiboSphere achieves strong performance in structure reconstruction (RMSD 1.25\,Å, TM-score 0.84), and its pretrained discrete representations transfer effectively to inverse folding and RNA--ligand binding prediction, with robust generalization in data-scarce regimes.
Lost in Tokenization: Context as the Key to Unlocking Biomolecular Understanding in Scientific LLMs
Oct 27, 2025Scientific Large Language Models (Sci-LLMs) have emerged as a promising frontier for accelerating biological discovery. However, these models face a fundamental challenge when processing raw biomolecular sequences: the tokenization dilemma. Whether treating sequences as a specialized language, risking the loss of functional motif information, or as a separate modality, introducing formidable alignment challenges, current strategies fundamentally limit their reasoning capacity. We challenge this sequence-centric paradigm by positing that a more effective strategy is to provide Sci-LLMs with high-level structured context derived from established bioinformatics tools, thereby bypassing the need to interpret low-level noisy sequence data directly. Through a systematic comparison of leading Sci-LLMs on biological reasoning tasks, we tested three input modes: sequence-only, context-only, and a combination of both. Our findings are striking: the context-only approach consistently and substantially outperforms all other modes. Even more revealing, the inclusion of the raw sequence alongside its high-level context consistently degrades performance, indicating that raw sequences act as informational noise, even for models with specialized tokenization schemes. These results suggest that the primary strength of existing Sci-LLMs lies not in their nascent ability to interpret biomolecular syntax from scratch, but in their profound capacity for reasoning over structured, human-readable knowledge. Therefore, we argue for reframing Sci-LLMs not as sequence decoders, but as powerful reasoning engines over expert knowledge. This work lays the foundation for a new class of hybrid scientific AI agents, repositioning the developmental focus from direct sequence interpretation towards high-level knowledge synthesis. The code is available at github.com/opendatalab-raise-dev/CoKE.
From Supervision to Exploration: What Does Protein Language Model Learn During Reinforcement Learning?
Oct 02, 2025Protein language models (PLMs) have advanced computational protein science through large-scale pretraining and scalable architectures. In parallel, reinforcement learning (RL) has broadened exploration and enabled precise multi-objective optimization in protein design. Yet whether RL can push PLMs beyond their pretraining priors to uncover latent sequence-structure-function rules remains unclear. We address this by pairing RL with PLMs across four domains: antimicrobial peptide design, kinase variant optimization, antibody engineering, and inverse folding. Using diverse RL algorithms and model classes, we ask if RL improves sampling efficiency and, more importantly, if it reveals capabilities not captured by supervised learning. Across benchmarks, RL consistently boosts success rates and sample efficiency. Performance follows a three-factor interaction: task headroom, reward fidelity, and policy capacity jointly determine gains. When rewards are accurate and informative, policies have sufficient capacity, and tasks leave room beyond supervised baselines, improvements scale; when rewards are noisy or capacity is constrained, gains saturate despite exploration. This view yields practical guidance for RL in protein design: prioritize reward modeling and calibration before scaling policy size, match algorithm and regularization strength to task difficulty, and allocate capacity where marginal gains are largest. Implementation is available at https://github.com/chq1155/RL-PLM.
DEL-Ranking: Ranking-Correction Denoising Framework for Elucidating Molecular Affinities in DNA-Encoded Libraries
Oct 19, 2024



DNA-encoded library (DEL) screening has revolutionized the detection of protein-ligand interactions through read counts, enabling rapid exploration of vast chemical spaces. However, noise in read counts, stemming from nonspecific interactions, can mislead this exploration process. We present DEL-Ranking, a novel distribution-correction denoising framework that addresses these challenges. Our approach introduces two key innovations: (1) a novel ranking loss that rectifies relative magnitude relationships between read counts, enabling the learning of causal features determining activity levels, and (2) an iterative algorithm employing self-training and consistency loss to establish model coherence between activity label and read count predictions. Furthermore, we contribute three new DEL screening datasets, the first to comprehensively include multi-dimensional molecular representations, protein-ligand enrichment values, and their activity labels. These datasets mitigate data scarcity issues in AI-driven DEL screening research. Rigorous evaluation on diverse DEL datasets demonstrates DEL-Ranking's superior performance across multiple correlation metrics, with significant improvements in binding affinity prediction accuracy. Our model exhibits zero-shot generalization ability across different protein targets and successfully identifies potential motifs determining compound binding affinity. This work advances DEL screening analysis and provides valuable resources for future research in this area.
Unlocking Potential Binders: Multimodal Pretraining DEL-Fusion for Denoising DNA-Encoded Libraries
Sep 07, 2024



In the realm of drug discovery, DNA-encoded library (DEL) screening technology has emerged as an efficient method for identifying high-affinity compounds. However, DEL screening faces a significant challenge: noise arising from nonspecific interactions within complex biological systems. Neural networks trained on DEL libraries have been employed to extract compound features, aiming to denoise the data and uncover potential binders to the desired therapeutic target. Nevertheless, the inherent structure of DEL, constrained by the limited diversity of building blocks, impacts the performance of compound encoders. Moreover, existing methods only capture compound features at a single level, further limiting the effectiveness of the denoising strategy. To mitigate these issues, we propose a Multimodal Pretraining DEL-Fusion model (MPDF) that enhances encoder capabilities through pretraining and integrates compound features across various scales. We develop pretraining tasks applying contrastive objectives between different compound representations and their text descriptions, enhancing the compound encoders' ability to acquire generic features. Furthermore, we propose a novel DEL-fusion framework that amalgamates compound information at the atomic, submolecular, and molecular levels, as captured by various compound encoders. The synergy of these innovations equips MPDF with enriched, multi-scale features, enabling comprehensive downstream denoising. Evaluated on three DEL datasets, MPDF demonstrates superior performance in data processing and analysis for validation tasks. Notably, MPDF offers novel insights into identifying high-affinity molecules, paving the way for improved DEL utility in drug discovery.
Deep Learning-Based Quasi-Conformal Surface Registration for Partial 3D Faces Applied to Facial Recognition
May 16, 2024



3D face registration is an important process in which a 3D face model is aligned and mapped to a template face. However, the task of 3D face registration becomes particularly challenging when dealing with partial face data, where only limited facial information is available. To address this challenge, this paper presents a novel deep learning-based approach that combines quasi-conformal geometry with deep neural networks for partial face registration. The proposed framework begins with a Landmark Detection Network that utilizes curvature information to detect the presence of facial features and estimate their corresponding coordinates. These facial landmark features serve as essential guidance for the registration process. To establish a dense correspondence between the partial face and the template surface, a registration network based on quasiconformal theories is employed. The registration network establishes a bijective quasiconformal surface mapping aligning corresponding partial faces based on detected landmarks and curvature values. It consists of the Coefficients Prediction Network, which outputs the optimal Beltrami coefficient representing the surface mapping. The Beltrami coefficient quantifies the local geometric distortion of the mapping. By controlling the magnitude of the Beltrami coefficient through a suitable activation function, the bijectivity and geometric distortion of the mapping can be controlled. The Beltrami coefficient is then fed into the Beltrami solver network to reconstruct the corresponding mapping. The surface registration enables the acquisition of corresponding regions and the establishment of point-wise correspondence between different partial faces, facilitating precise shape comparison through the evaluation of point-wise geometric differences at these corresponding regions. Experimental results demonstrate the effectiveness of the proposed method.
An Autonomous Large Language Model Agent for Chemical Literature Data Mining
Feb 20, 2024



Chemical synthesis, which is crucial for advancing material synthesis and drug discovery, impacts various sectors including environmental science and healthcare. The rise of technology in chemistry has generated extensive chemical data, challenging researchers to discern patterns and refine synthesis processes. Artificial intelligence (AI) helps by analyzing data to optimize synthesis and increase yields. However, AI faces challenges in processing literature data due to the unstructured format and diverse writing style of chemical literature. To overcome these difficulties, we introduce an end-to-end AI agent framework capable of high-fidelity extraction from extensive chemical literature. This AI agent employs large language models (LLMs) for prompt generation and iterative optimization. It functions as a chemistry assistant, automating data collection and analysis, thereby saving manpower and enhancing performance. Our framework's efficacy is evaluated using accuracy, recall, and F1 score of reaction condition data, and we compared our method with human experts in terms of content correctness and time efficiency. The proposed approach marks a significant advancement in automating chemical literature extraction and demonstrates the potential for AI to revolutionize data management and utilization in chemistry.
A Survey on Generative Diffusion Model
Sep 21, 2022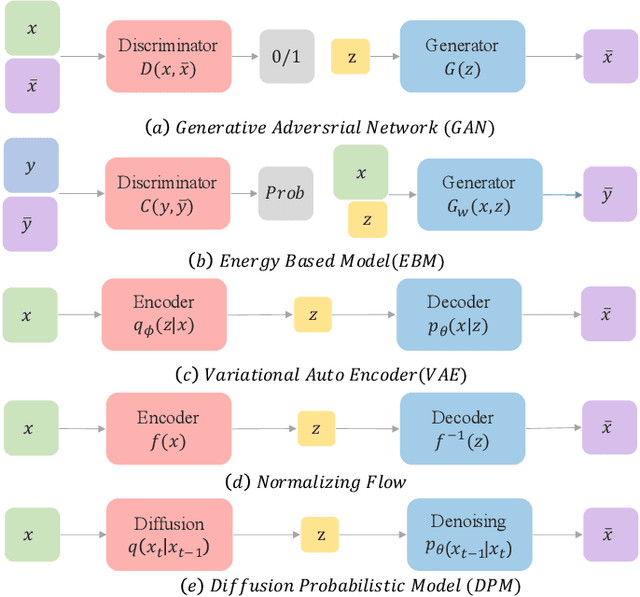
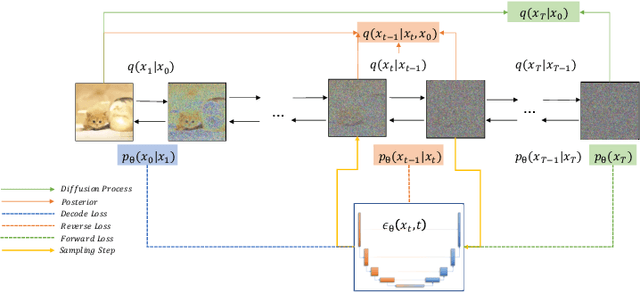
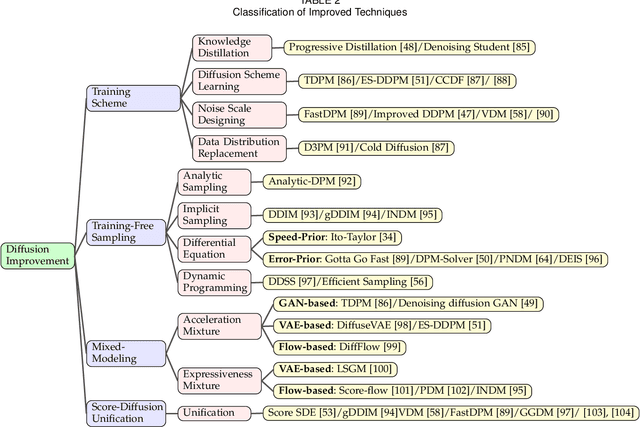
Deep learning shows great potential in generation tasks thanks to deep latent representation. Generative models are classes of models that can generate observations randomly with respect to certain implied parameters. Recently, the diffusion Model becomes a raising class of generative models by virtue of its power-generating ability. Nowadays, great achievements have been reached. More applications except for computer vision, speech generation, bioinformatics, and natural language processing are to be explored in this field. However, the diffusion model has its natural drawback of a slow generation process, leading to many enhanced works. This survey makes a summary of the field of the diffusion model. We firstly state the main problem with two landmark works - DDPM and DSM. Then, we present a diverse range of advanced techniques to speed up the diffusion models - training schedule, training-free sampling, mixed-modeling, and score & diffusion unification. Regarding existing models, we also provide a benchmark of FID score, IS, and NLL according to specific NFE. Moreover, applications with diffusion models are introduced including computer vision, sequence modeling, audio, and AI for science. Finally, there is a summarization of this field together with limitations & further directions.