 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeOpenChemIE: An Information Extraction Toolkit For Chemistry Literature
Apr 01, 2024



Information extraction from chemistry literature is vital for constructing up-to-date reaction databases for data-driven chemistry. Complete extraction requires combining information across text, tables, and figures, whereas prior work has mainly investigated extracting reactions from single modalities. In this paper, we present OpenChemIE to address this complex challenge and enable the extraction of reaction data at the document level. OpenChemIE approaches the problem in two steps: extracting relevant information from individual modalities and then integrating the results to obtain a final list of reactions. For the first step, we employ specialized neural models that each address a specific task for chemistry information extraction, such as parsing molecules or reactions from text or figures. We then integrate the information from these modules using chemistry-informed algorithms, allowing for the extraction of fine-grained reaction data from reaction condition and substrate scope investigations. Our machine learning models attain state-of-the-art performance when evaluated individually, and we meticulously annotate a challenging dataset of reaction schemes with R-groups to evaluate our pipeline as a whole, achieving an F1 score of 69.5%. Additionally, the reaction extraction results of \ours attain an accuracy score of 64.3% when directly compared against the Reaxys chemical database. We provide OpenChemIE freely to the public as an open-source package, as well as through a web interface.
Beyond Major Product Prediction: Reproducing Reaction Mechanisms with Machine Learning Models Trained on a Large-Scale Mechanistic Dataset
Mar 07, 2024



Mechanistic understanding of organic reactions can facilitate reaction development, impurity prediction, and in principle, reaction discovery. While several machine learning models have sought to address the task of predicting reaction products, their extension to predicting reaction mechanisms has been impeded by the lack of a corresponding mechanistic dataset. In this study, we construct such a dataset by imputing intermediates between experimentally reported reactants and products using expert reaction templates and train several machine learning models on the resulting dataset of 5,184,184 elementary steps. We explore the performance and capabilities of these models, focusing on their ability to predict reaction pathways and recapitulate the roles of catalysts and reagents. Additionally, we demonstrate the potential of mechanistic models in predicting impurities, often overlooked by conventional models. We conclude by evaluating the generalizability of mechanistic models to new reaction types, revealing challenges related to dataset diversity, consecutive predictions, and violations of atom conservation.
Substrate Scope Contrastive Learning: Repurposing Human Bias to Learn Atomic Representations
Feb 19, 2024



Learning molecular representation is a critical step in molecular machine learning that significantly influences modeling success, particularly in data-scarce situations. The concept of broadly pre-training neural networks has advanced fields such as computer vision, natural language processing, and protein engineering. However, similar approaches for small organic molecules have not achieved comparable success. In this work, we introduce a novel pre-training strategy, substrate scope contrastive learning, which learns atomic representations tailored to chemical reactivity. This method considers the grouping of substrates and their yields in published substrate scope tables as a measure of their similarity or dissimilarity in terms of chemical reactivity. We focus on 20,798 aryl halides in the CAS Content Collection spanning thousands of publications to learn a representation of aryl halide reactivity. We validate our pre-training approach through both intuitive visualizations and comparisons to traditional reactivity descriptors and physical organic chemistry principles. The versatility of these embeddings is further evidenced in their application to yield prediction, regioselectivity prediction, and the diverse selection of new substrates. This work not only presents a chemistry-tailored neural network pre-training strategy to learn reactivity-aligned atomic representations, but also marks a first-of-its-kind approach to benefit from the human bias in substrate scope design.
Predictive Chemistry Augmented with Text Retrieval
Dec 08, 2023



This paper focuses on using natural language descriptions to enhance predictive models in the chemistry field. Conventionally, chemoinformatics models are trained with extensive structured data manually extracted from the literature. In this paper, we introduce TextReact, a novel method that directly augments predictive chemistry with texts retrieved from the literature. TextReact retrieves text descriptions relevant for a given chemical reaction, and then aligns them with the molecular representation of the reaction. This alignment is enhanced via an auxiliary masked LM objective incorporated in the predictor training. We empirically validate the framework on two chemistry tasks: reaction condition recommendation and one-step retrosynthesis. By leveraging text retrieval, TextReact significantly outperforms state-of-the-art chemoinformatics models trained solely on molecular data.
Pareto Optimization to Accelerate Multi-Objective Virtual Screening
Oct 16, 2023The discovery of therapeutic molecules is fundamentally a multi-objective optimization problem. One formulation of the problem is to identify molecules that simultaneously exhibit strong binding affinity for a target protein, minimal off-target interactions, and suitable pharmacokinetic properties. Inspired by prior work that uses active learning to accelerate the identification of strong binders, we implement multi-objective Bayesian optimization to reduce the computational cost of multi-property virtual screening and apply it to the identification of ligands predicted to be selective based on docking scores to on- and off-targets. We demonstrate the superiority of Pareto optimization over scalarization across three case studies. Further, we use the developed optimization tool to search a virtual library of over 4M molecules for those predicted to be selective dual inhibitors of EGFR and IGF1R, acquiring 100% of the molecules that form the library's Pareto front after exploring only 8% of the library. This workflow and associated open source software can reduce the screening burden of molecular design projects and is complementary to research aiming to improve the accuracy of binding predictions and other molecular properties.
Learning Over Molecular Conformer Ensembles: Datasets and Benchmarks
Sep 29, 2023



Molecular Representation Learning (MRL) has proven impactful in numerous biochemical applications such as drug discovery and enzyme design. While Graph Neural Networks (GNNs) are effective at learning molecular representations from a 2D molecular graph or a single 3D structure, existing works often overlook the flexible nature of molecules, which continuously interconvert across conformations via chemical bond rotations and minor vibrational perturbations. To better account for molecular flexibility, some recent works formulate MRL as an ensemble learning problem, focusing on explicitly learning from a set of conformer structures. However, most of these studies have limited datasets, tasks, and models. In this work, we introduce the first MoleculAR Conformer Ensemble Learning (MARCEL) benchmark to thoroughly evaluate the potential of learning on conformer ensembles and suggest promising research directions. MARCEL includes four datasets covering diverse molecule- and reaction-level properties of chemically diverse molecules including organocatalysts and transition-metal catalysts, extending beyond the scope of common GNN benchmarks that are confined to drug-like molecules. In addition, we conduct a comprehensive empirical study, which benchmarks representative 1D, 2D, and 3D molecular representation learning models, along with two strategies that explicitly incorporate conformer ensembles into 3D MRL models. Our findings reveal that direct learning from an accessible conformer space can improve performance on a variety of tasks and models.
Artificial Intelligence for Science in Quantum, Atomistic, and Continuum Systems
Jul 17, 2023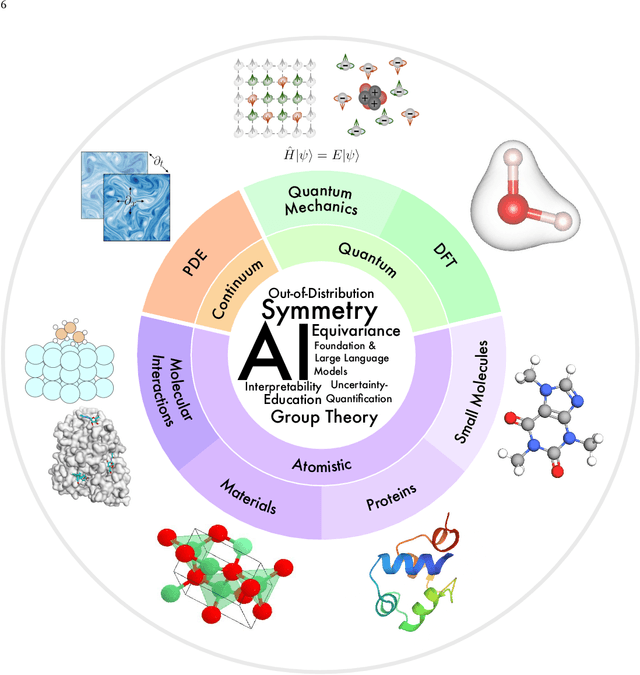



Advances in artificial intelligence (AI) are fueling a new paradigm of discoveries in natural sciences. Today, AI has started to advance natural sciences by improving, accelerating, and enabling our understanding of natural phenomena at a wide range of spatial and temporal scales, giving rise to a new area of research known as AI for science (AI4Science). Being an emerging research paradigm, AI4Science is unique in that it is an enormous and highly interdisciplinary area. Thus, a unified and technical treatment of this field is needed yet challenging. This paper aims to provide a technically thorough account of a subarea of AI4Science; namely, AI for quantum, atomistic, and continuum systems. These areas aim at understanding the physical world from the subatomic (wavefunctions and electron density), atomic (molecules, proteins, materials, and interactions), to macro (fluids, climate, and subsurface) scales and form an important subarea of AI4Science. A unique advantage of focusing on these areas is that they largely share a common set of challenges, thereby allowing a unified and foundational treatment. A key common challenge is how to capture physics first principles, especially symmetries, in natural systems by deep learning methods. We provide an in-depth yet intuitive account of techniques to achieve equivariance to symmetry transformations. We also discuss other common technical challenges, including explainability, out-of-distribution generalization, knowledge transfer with foundation and large language models, and uncertainty quantification. To facilitate learning and education, we provide categorized lists of resources that we found to be useful. We strive to be thorough and unified and hope this initial effort may trigger more community interests and efforts to further advance AI4Science.
RxnScribe: A Sequence Generation Model for Reaction Diagram Parsing
May 19, 2023Reaction diagram parsing is the task of extracting reaction schemes from a diagram in the chemistry literature. The reaction diagrams can be arbitrarily complex, thus robustly parsing them into structured data is an open challenge. In this paper, we present RxnScribe, a machine learning model for parsing reaction diagrams of varying styles. We formulate this structured prediction task with a sequence generation approach, which condenses the traditional pipeline into an end-to-end model. We train RxnScribe on a dataset of 1,378 diagrams and evaluate it with cross validation, achieving an 80.0% soft match F1 score, with significant improvements over previous models. Our code and data are publicly available at https://github.com/thomas0809/RxnScribe.
Prefix-tree Decoding for Predicting Mass Spectra from Molecules
Mar 11, 2023



Computational predictions of mass spectra from molecules have enabled the discovery of clinically relevant metabolites. However, such predictive tools are still limited as they occupy one of two extremes, either operating (a) by fragmenting molecules combinatorially with overly rigid constraints on potential rearrangements and poor time complexity or (b) by decoding lossy and nonphysical discretized spectra vectors. In this work, we introduce a new intermediate strategy for predicting mass spectra from molecules by treating mass spectra as sets of chemical formulae, which are themselves multisets of atoms. After first encoding an input molecular graph, we decode a set of chemical subformulae, each of which specify a predicted peak in the mass spectra, the intensities of which are predicted by a second model. Our key insight is to overcome the combinatorial possibilities for chemical subformulae by decoding the formula set using a prefix tree structure, atom-type by atom-type, representing a general method for ordered multiset decoding. We show promising empirical results on mass spectra prediction tasks.
Reinforced Genetic Algorithm for Structure-based Drug Design
Nov 28, 2022


Structure-based drug design (SBDD) aims to discover drug candidates by finding molecules (ligands) that bind tightly to a disease-related protein (targets), which is the primary approach to computer-aided drug discovery. Recently, applying deep generative models for three-dimensional (3D) molecular design conditioned on protein pockets to solve SBDD has attracted much attention, but their formulation as probabilistic modeling often leads to unsatisfactory optimization performance. On the other hand, traditional combinatorial optimization methods such as genetic algorithms (GA) have demonstrated state-of-the-art performance in various molecular optimization tasks. However, they do not utilize protein target structure to inform design steps but rely on a random-walk-like exploration, which leads to unstable performance and no knowledge transfer between different tasks despite the similar binding physics. To achieve a more stable and efficient SBDD, we propose Reinforced Genetic Algorithm (RGA) that uses neural models to prioritize the profitable design steps and suppress random-walk behavior. The neural models take the 3D structure of the targets and ligands as inputs and are pre-trained using native complex structures to utilize the knowledge of the shared binding physics from different targets and then fine-tuned during optimization. We conduct thorough empirical studies on optimizing binding affinity to various disease targets and show that RGA outperforms the baselines in terms of docking scores and is more robust to random initializations. The ablation study also indicates that the training on different targets helps improve performance by leveraging the shared underlying physics of the binding processes. The code is available at https://github.com/futianfan/reinforced-genetic-algorithm.